
Los síndromes de neoplasia endocrina múltiple tipo 1 y tipo 2 (NEM1 y 2) constituyen un grupo heterogéneo de tumores que afectan glándulas endocrinas y no-endocrinas (1). NEM1 y 2 constituyen trastornos hereditarios raros que se transmiten en forma autosómica dominante y son causados por mutaciones en el gen MEN1 (2) y en el protooncogen RET (3), respectivamente.
NEM1.
En la actualidad se han descrito más de 20 combinaciones posibles, entre tumores endocrinos y no-endocrinos, asociados a NEM1. Los tumores endocrinos más comúnmente observados en NEM1 afectan a la glándula paratiroides (hiperparatiroidismo), páncreas endocrino (gastrinoma, insulinoma, glucagonoma) e hipófisis anterior (prolactinoma). Dentro de los tumores no-endocrinos asociados se hallan los tumores carcinoides (lipomas, angiofibromas faciales, colagenomas). En menor frecuencia, se halla asociado a tumores adrenocorticales (hipercortisolismo y hiperaldosteronismo primario). Debido al amplio espectro de combinaciones tumorales posibles, el diagnóstico de NEM1 no es sencillo de realizar. NEM1 puede diagnosticarse bajo la forma familiar o esporádica. Los estudios genéticos se enfocan inicialmente a la identificación de mutaciones puntuales mediante secuenciación completa del gen MEN1 (80-90% casos familiares vs. 65% casos esporádicos). De no hallarse mutaciones asociadas en esta primera etapa, se recomienda realizar la detección de grandes duplicaciones y/o deleciones (1-4% casos familiares) (4). Es importante tener presente que la posibilidad de hallar mutaciones causales en el gen MEN1 es poco probable para aquellos casos esporádicos que presenten sólo un único tumor asociado a MEN1 (1).
NEM2.
El carcinoma medular de tiroides (CMT) es un tumor maligno poco frecuente, originado a partir de células parafoliculares tipo C, las cuales tiene por función regular el metabolismo del calcio. Tiene una baja frecuencia, representando un 5% de los carcinomas tiroideos (1). El CMT puede ocurrir en forma esporádica (75%), o en forma hereditaria (25%), esta última en el contexto de un CMT familiar (CMTF) o de una neoplasia endocrina múltiple tipo 2A ó 2B (NEM 2A, 2B) (5). En NEM 2A, el CMT se halla asociado a feocromocitoma e hiperparatiroidismo, mientras que en NEM 2B, se lo puede hallar asociado a feocromocitoma y neuromas (cutaneomucosos y gastrointestinales). La mayoría de las mutaciones asociadas a NEM2A se hallan localizadas en los exones 10, 11 y 14 del gen RET (95%); mientras que aquellas asociadas a NEM2B se localizan en su mayoría en el exón 16 (95%). Alrededor del 50% de los pacientes con CMTF presentan mutaciones en los exones 13, 14 y 16. El diagnóstico precoz diferencial de CMT es esencial debido a la prognosis y a las diferentes estrategias terapéuticas disponibles (3).
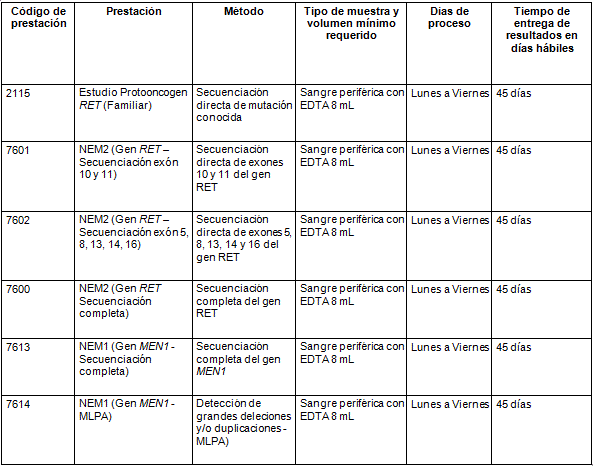
Determinaciones disponibles en Cibic:
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Bibliografía
1. Brandi y cols. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J clini endocrinol Metab. 2001, 86:5658-71.
2. Agarwal y cols .Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet. 1997, 6:1169-75.
3. Sanso y cols. Very early detection of RET protooncogene mutation is crucial for preventive thyroidectomy in multiple endocrine neoplasia type 2 children. Cancer 2002; 94:323-30.
4. Ellard y cols. Detection of an MEN1 gene mutation depends on clinical features and supports current referral criteria for diagnostic molecular genetic testing. Clin Endocrinol (Oxf) 2005; 62:169-75.
5. Farndon y cols. Familial medullary thyroid carcinoma without associated endocrino- pathies: a distinct clinical entity. Brit. J. Surg. 1986; 73: 278-81.
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Analía Seravalle Tel: 0341 4499444 Int: 242
Dra. María Florencia Gosso Tel: 0341 4499444 Int: 258