

Cada 8 de septiembre se conmemora el Día Mundial de la Fibrosis Quística, una jornada clave para visibilizar una enfermedad que, aunque poco frecuente, tiene un gran impacto en quienes conviven con ella.
Según el Registro Nacional de Fibrosis Quística (ReNaFQ), en Argentina hay entre 1.500 y 2.000 pacientes diagnosticados, y cada año nacen aproximadamente 100 niños con la patología. A pesar de los avances en diagnóstico y tratamiento, entre un 10% y 15% de los casos recién se detectan en la adultez, debido a subregistro y subdiagnóstico, según reporta el ReNaFQ.
A nivel mundial, se estima que existen alrededor de 170.000 personas con fibrosis quística, con un diagnóstico confirmado en aproximadamente el 65% a 70% de los casos.
Entendiendo la enfermedad: El Gen, la Proteína y el Moco
La enfermedad afecta a las células epiteliales exócrinas y se caracteriza por la producción de un moco espeso y viscoso, que obstruye los conductos de diversos órganos, principalmente pulmones y páncreas.
El gen responsable de la FQ (CFTR) comprende 27 exones y codifica una proteína de 1480 aminoácidos (CFTR, Cystic Fibrosis Transmembrane Conductance Regulator), localizada en la membrana apical de las células epiteliales. Esta proteína actúa como canal de cloro regulador del transporte iónico.
Se han descrito más de 2000 variantes en el gen CFTR, de las cuales varias son claramente patogénicas. La variante patogénica más común es la deleción de una fenilalanina en la posición 508 (p.Phe508del o F508del), presente en alrededor del 70% de los alelos en poblaciones de Europa del Norte, aunque con frecuencias menores en otras regiones del mundo, como América Latina.
El Diagnóstico: Un Camino que Empieza con una Sospecha
El diagnóstico de la fibrosis quística (FQ) se fundamenta en la combinación de criterios clínicos, antecedentes familiares, pesquisa neonatal y estudios de laboratorio.
1 – Sospecha clínica
Debe considerarse la posibilidad de FQ cuando se presenta una o más de las siguientes condiciones:
• Enfermedad respiratoria o sinusopulmonar crónica.
• Alteraciones gastrointestinales o nutricionales (insuficiencia pancreática, malabsorción, desnutrición).
• Síndrome de pérdida excesiva de sal.
• Azoospermia obstructiva.
• Hermano/a diagnosticado con FQ.
• Pesquisa neonatal positiva para FQ.
2 – Confirmación diagnóstica
La enfermedad se confirma mediante alguno de los siguientes criterios:
• Prueba de sudor positiva: concentración de cloro ≥ 60 mmol/L en dos determinaciones independientes. Valores intermedios (30–59 mmol/L en niños; 40–59 mmol/L en adultos) requieren estudios adicionales.
• Estudio genético: identificación de dos variantes patogénicas en el gen CFTR (al tratarse de una patología autosómica recesiva).
• Pruebas funcionales en casos dudosos: demostración de alteración en la diferencia de potencial transepitelial nasal o en la medición de la corriente intestinal (ICM), disponibles en centros especializados.
Es importante destacar que la pesquisa neonatal positiva constituye un hallazgo de sospecha, pero siempre debe confirmarse con prueba de sudor y/o análisis genético antes de establecer el diagnóstico definitivo.
Desde la identificación del gen CFTR, se han descripto casos de pacientes con variantes infrecuentes que pueden presentar manifestaciones clínicas atípicas e incluso valores normales en la prueba del sudor. Por este motivo, las guías actuales recomiendan que el diagnóstico de fibrosis quística se realice cuando existe al menos un criterio clínico compatible o pesquisa neonatal positiva, en conjunto con la demostración de disfunción del canal CFTR, ya sea mediante concentración de cloro en sudor ≥ 60 mmol/L, la identificación de dos variantes patogénicas o probablemente patogénicas en el gen CFTR, o pruebas funcionales de transporte iónico anormales.
Una Revolución Terapéutica y el marco Legal en Argentina
En los últimos años el enfoque terapéutico está cambiando desde un tratamiento sintomático hacia uno dirigido al defecto genético. Esto consiste en corregir la alteración genética del paciente o lograr que la proteína alterada funcione normalmente. El objetivo es prevenir, tratar o reducir las exacerbaciones y el déficit nutricional, para así mejorar la calidad de vida de los pacientes.
Los fármacos que modulan la proteína reguladora de la conductancia transmembrana de la fibrosis quística (CFTR), concretamente ivacaftor, lumacaftor, tezacaftor y elexacaftor, están revolucionando el tratamiento de los pacientes con fibrosis quística (FQ), en particular aquellos con al menos una variante F508del (hasta el 85 % de los pacientes).
Desde el 2021 nuestro país cuenta con la fabricación nacional de la triple terapia moduladora (ivacaftor, lumacaftor, tezacaftor) con aprobación de la ANMAT, con indicaciones específicas para determinadas variantes genéticas abarcando un alto porcentaje de pacientes.
En Argentina, la Ley 24438, sancionada en 1994, incluyó la fibrosis quística en el tamizaje neonatal junto con otras afecciones congénitas. Esta medida representó un paso importante, ya que posibilitó la detección de la enfermedad antes de los dos meses de vida y la puesta en marcha de tratamientos tempranos. Sin embargo, se calcula que entre el 10% y el 15% de los casos aún se identifican en la adultez.
En 2020, la Ley 27552 otorgó un marco legal nacional a los derechos de las personas con fibrosis quística. A partir de esta normativa, la enfermedad quedó comprendida dentro del programa de enfermedades poco frecuentes, lo que garantiza cobertura y acceso a servicios de salud, educación, empleo, rehabilitación, seguridad social y actividades de prevención para los pacientes de cualquier edad.
Nuestro Compromiso desde el Laboratorio
Para acompañar cada paso del diagnóstico de la Fibrosis Quística, en nuestro laboratorio ofrecemos un panel completo de prestaciones que van desde el screening hasta la confirmación genética de alta complejidad.
Prestaciones disponibles en Cibic:
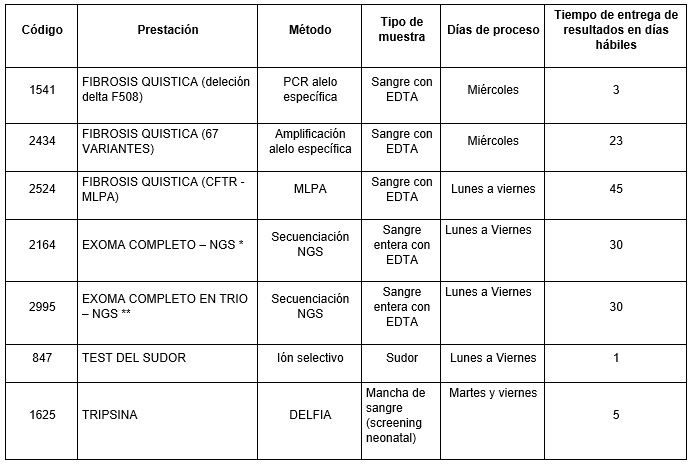
* Exoma completo dirigido a patología específica con diagnóstico presuntivo y/o a la sintomatología del paciente de acuerdo a términos HPO.
Puede solicitarse adicionalmente el estudio del genoma mitocondrial cuando hay compromiso neuromuscular, respiratorio y cuando se sospecha participación de la mitocondria en el desarrollo de la enfermedad,
** Exoma completo trío incluye a los padres del paciente para mejorar el rinde diagnóstico cuando sea necesario
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Referencias:
– Registro Nacional de Fibrosis Quística (ReNaFQ).
Sitio web: https://www.fibrosisquistica.org.ar
– Sociedad Argentina de Pediatría (SAP).
“Guía para el diagnóstico y tratamiento de la fibrosis quística en Argentina”.
Disponible en: https://www.sap.org.ar
– Leyes argentinas relacionadas con FQ:
Ley 24438 (1994) – Inclusión de fibrosis quística en tamizaje neonatal.
Ley 27552 (2020) – Derechos de las personas con fibrosis quística
– Cystic Fibrosis Foundation (CFF).
Sitio web: https://www.cff.org
Datos globales, tratamientos innovadores y estadísticas sobre prevalencia y diagnóstico.
– European Cystic Fibrosis Society (ECFS).
Sitio web: https://www.ecfs.eu
– Ratjen F, Döring G. Cystic fibrosis. Lancet. 2003;361:681–689.
– Elborn JS. Cystic fibrosis. Lancet. 2016;388:2519–2531.
– De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med. 2016;4(8):662–674.
– Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015;16(1):45–56.
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Alan Gomez
Tel: 0341-486-1600. Interno: 225
Sección: Producción bioquímica clínica
Bioq. María Eugenia Dacharry
Tel: 0341-486-1600. Interno: 225