

El Síndrome Mielodisplásico (SMD) es un grupo de trastornos clonales de células madre hematopoyéticas caracterizados por citopenia periférica, progenitores hematopoyéticos displásicos, médula ósea hipercelular y alto riesgo de conversión a Leucemia Mieloide Aguda (LMA). Los SMD ocurren principalmente en adultos mayores, con una edad media al diagnóstico que está entre los 60 y 75 años. Con respecto al sexo, predomina en hombres (1).
Los síntomas son atribuibles a la línea celular específica más afectada y pueden consistir en: cansancio, debilidad, palidez (secundaria a anemia), aumento de infecciones y fiebre (secundarias a neutropenia) y aumento de hemorragia y hematomas (secundario a trombocitopenia).
Para el diagnostico, la displasia debe comprometer al menos el 10% de alguna de las líneas mieloides y el conteo de blastos debe ser <20% (valor a partir del cual debe considerarse el diagnóstico de LMA), pero se establece al demostrar anormalidades citogenéticas específicas y mutaciones somáticas. Puede producirse una hipoplasia de la medula ósea.
La etiología de los síndromes mielodisplásicos se desconoce. El riesgo aumenta con la edad debido a la adquisición de mutaciones somáticas que pueden promover la expansión clonal y el predominio de una célula madre hematopoyética específica, y posiblemente debido a la exposición a toxinas ambientales como benceno, radiación y agentes antineoplásicos (en particular, esquemas prolongados o intensos, y los que incorporan agentes alquilantes, hidroxiurea y/o inhibidores de la topoisomerasa) (2).
Las aberraciones cromosómicas (ej.: deleciones, duplicaciones, anomalías estructurales) están presentes en la mitad de todos los pacientes con SMD de novo, y la frecuencia de aberración cromosómica varía con los cambios geográficos y étnicos. Varias anomalías citogenéticas en los SMD se observan también en la LMA, lo que respalda un origen común para una proporción de estas dos enfermedades.
En los síndromes mielodisplásicos están presentes alteraciones citogenéticas frecuentes como la deleción de los cromosomas 5q, 7q y 20q, la monosomía del cromosoma 7, la trisomía del cromosoma 8 y la presencia de cariotipos complejos, que, unido a mutaciones somáticas en diferentes genes, intervienen en la patogénesis de la enfermedad y su conocimiento permite la estratificación pronóstica de los pacientes (3).
La monosomía 5 o deleción del brazo largo del cromosoma 5 (5q-) fue la anomalía cromosómica más frecuente encontrada en todo el mundo y tienen un pronóstico favorable.
La monosomía 7 se caracteriza por citopenias refractarias graves y susceptibilidad a infecciones importantes y se considera un predictor independiente de supervivencia en pacientes con alto riesgo.
En cuanto a la trisomía del cromosoma 8, aparece tardíamente durante el curso de la enfermedad. Desde el punto de vista clínico está asociada a manifestaciones de autoinmunidad; por lo que los pacientes responden de forma satisfactoria a terapias inmunosupresoras.
El cromosoma 17 puede presentarse como delecion 17p, o bien, como isocromosoma 17q en SMD. Los pacientes con isocromosoma 17q se caracterizan por anemia grave, leucocitosis con neutrófilos, anomalía pseudo Pelger- Huët e hiperplasia de la médula ósea.
La aparición de cariotipos complejos es frecuente en los SMD e incluye al menos tres o más aberraciones que pueden ser tanto numéricas como estructurales. Estos casos reflejan una inestabilidad cromosómica inherente que contribuye a la progresión de la enfermedad y peor pronóstico.
En Cibic Laboratorios, a través de la prestación 2466 disponemos de un panel para SMD que cuenta con las siguientes sondas (las cuales también puede ser requeridas de forma individual):
• 5q31(EGR1)
• 7q31(MET)
• Centrómero cromosoma 8
• 17p13(TP53)
Prestaciones disponibles en Cibic Laboratorios:
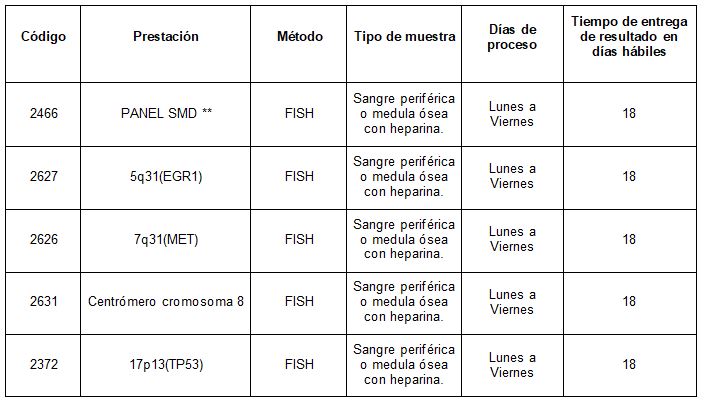
** PANEL SMD: 5q31(EGR1), 7q31(MET), Centrómero cromosoma 8, 17p13(TP53)
Referencias:
1. Cataño Pulgarin, J. C., Franco, O. A. & Orduz, Y. R. Síndrome mielodisplásico: aspectos básicos y abordaje diagnóstico. Rev. Colomb. Hematol. Oncol. 8, 90–106 (2021).
2. Zahid, M. F. et al. Cytogenetic Abnormalities in Myelodysplastic Syndromes: An Overview. Int. J. Hematol.-Oncol. Stem Cell Res. 11, 231–239 (2017).
3. The Principles of Clinical Cytogenetics. (Springer, 2013). doi:10.1007/978-1-4419-1688-4
Para mayor información o consultas:
Sección: Citogenética
Bioq. Sibila Bertalot
Lic. Veronica Vanrell
Téc. Victorina Carbone
Téc. Eliana Navilli
Tel: 0341-4861600. Int: 283