

El Síndrome Linfoproliferativo Autoinmune (ALPS) es una rara condición generada por la alteración de los mecanismos de apoptosis de linfocitos T que perturban la homeostasia linfocitaria. Se define como un trastorno linfoproliferativo no maligno hereditario caracterizado por variantes patogénicas en genes relevantes dentro de la vía de señalización del receptor de señal de apoptosis (FAS) como lo son FAS, CASP10 y FASGL.
Identificado inicialmente en la década de 1990, se observó ALPS en una cohorte de pacientes con enfermedad linfoproliferativa crónica y que, además, presentaban un mayor número de una población de células T característica denominada “células T doble negativas” (DNT) (1). Defectos en la apoptosis mediada por FAS causan una expansión y acumulación de células T autorreactivas que expresan CD3 pero son CD4 y CD8 doble negativas, lo que produce citopenias, esplenomegalia, linfadenopatía, trastornos autoinmunes y un mayor riesgo de por vida de padecer linfoma.
En condiciones normales, la activación de las células T induce la expresión de FASL, que puede unirse a los receptores FAS en la misma célula o en células cercanas. Este evento resulta en la agrupación de receptores FAS y en la unión de proteínas asociadas a FAS con el dominio de muerte (FADD). El FADD luego recluta a la pro-caspasa 8 y pro-caspasa 10 para formar el complejo de señalización que induce la muerte celular (DISC), el cual propaga una señal que conduce a la activación de la caspasa terminal y produce finalmente la apoptosis. Este proceso es conocido como muerte celular inducida por activación (AICD) (2).
En individuos sanos, las células T periféricas que reaccionan con los autoantígenos sufren estimulación repetida, lo que conduce a un aumento de la producción de FASL, a la formación de DISC y finalmente a AICD. Este sistema previene la expansión de las células T autorreactivas, lo que evita que se puedan desencadenar enfermedades autoinmunes. En cambio, los pacientes con ALPS tienen variantes patogénicas en miembros de la vía FAS, lo que hace que sus células T sean defectuosas en AICD.
Los pacientes con ALPS acumulan células T autorreactivas, que clásicamente son negativas tanto para CD4 como para CD8, y positivas para el receptor de células T Alfa/Beta (TCR) (2). El reconocimiento de ALPS es fundamental, ya que el tratamiento con terapias inmunosupresoras puede reducir o mejorar eficazmente los síntomas en la mayoría de los pacientes.
El estudio de un grupo de 250 pacientes con ALPS mostró que la mayoría presentaba linfadenopatía (>90%) y esplenomegalia (95%). La mayor parte de los pacientes desarrollaron linfoproliferación a una edad temprana (mediana 11,5 meses). La linfoproliferación tendió a mejorar con la edad (en edad adulta) y se resolvió en la mayoría de los pacientes. La citopenia autoinmune (53%) puede ser la primera manifestación de la enfermedad en ausencia de linfoproliferación. La anemia hemolítica autoinmune (33%) es el evento más frecuente. Por lo tanto, cualquier paciente con bicitopenia o tricitopenia autoinmune debe realizarse pruebas para ALPS (3).
Además de los síntomas hematológicos, se han descrito otros signos de autoinmunidad, como lesiones cutáneas eccematosas, hepatitis autoinmune, vasculitis, uveítis, tiroiditis autoinmune y glomerulonefritis. Los pacientes ALPS también tienen un mayor riesgo de neoplasias malignas secundarias, más comúnmente Linfoma de Hodgkin y no Hodgkin. Se estima que el riesgo es entre 60 y 150 veces mayor que el de la población general (3).
Si bien existe hasta un 20 % de personas con ALPS clínico en las cuales no se ha identificado una etiología genética, en hasta el 80 % de los casos se han identificado variantes patogénicas en los genes FAS (de ALPS-FAS: es el gen mayormente afectado, tanto variantes somáticas como de línea germinal), CASP10 (ALPS-CASP10) y FASGL (ALPS-FASLG).
La identificación de variantes de línea germinal en pacientes afectados es de suma relevancia para el manejo del paciente y del grupo familiar. La herencia de ALPS-CASP10, la mayoría de los casos de ALPS-FAS y algunos casos de ALPS-FASLG es autosómica dominante. Cada hijo de un individuo con ALPS autosómico dominante tiene un 50% de posibilidades de heredar la variante patogénica. La herencia de la mayoría de los casos de ALPS-FASLG y ALPS grave asociado con variantes patogénicas bialélicas de FAS es autosómica recesiva. Es probable que los padres de un individuo con ALPS autosómico recesivo sean heterocigotos, en cuyo caso cada uno tiene una variante patogénica de FAS; estos padres pueden tener hallazgos relacionados con ALPS o pueden ser clínicamente asintomáticos (4).
La técnica de Citometría de Flujo permite la tipificación de los linfocitos T de sangre periférica. El análisis de la subpoblación de células T CD45+, CD3+ debe mostrar un subconjunto diferencial de CD4- y CD8- que representa a las células T defectuosas doble negativas (Figura 1, A y B). La tipificación de TCR de la población doble negativa que muestra una expresión restringida de T Alfa/Beta respalda firmemente el diagnóstico de ALPS (Figura 1,C) (2).
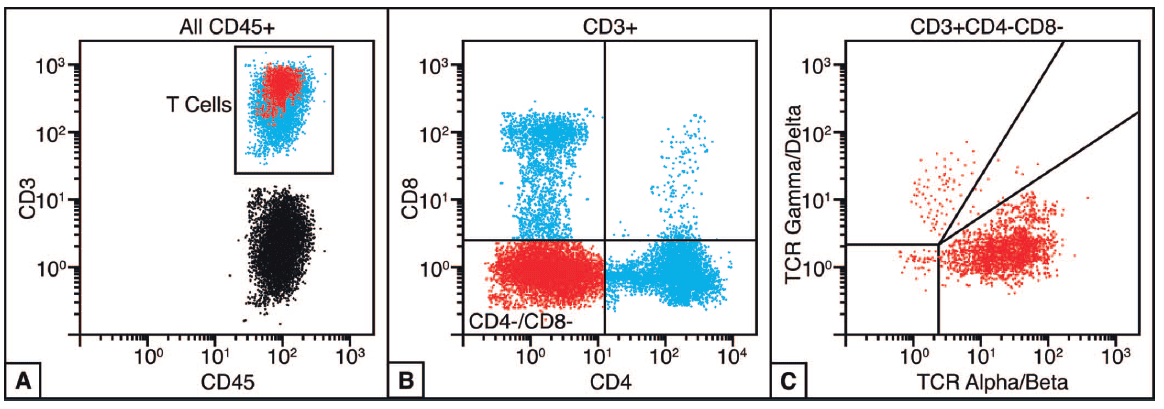
Figura 1. Resultados característicos de Citometría de Flujo en el Síndrome Linfoproliferativo Autoinmune.
Figuras A y B: La sangre periférica de pacientes con ALPS mostrarán un grupo de células T CD3+ que son negativas para CD4 y CD8 (B, en rojo).
Figura C: La población de células doble negativas debe ser positiva para el receptor T Alfa/Beta (TCR) (2).
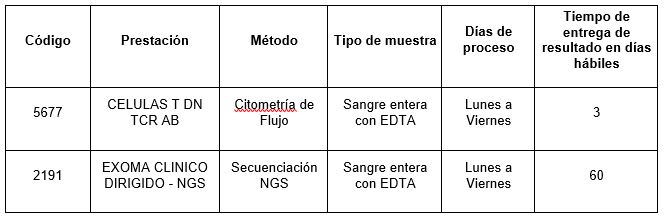
En Cibic Laboratorios disponemos de las siguientes prestaciones:
Referencias:
1. Bride, K. & Teachey, D. Autoimmune lymphoproliferative syndrome: more than a FAScinating disease. F1000Research 6, 1928 (2017).
2. Matson, D. R. & Yang, D. T. Autoimmune Lymphoproliferative Syndrome: An Overview. Arch. Pathol. Lab. Med. 144, 245–251 (2020).
3. Casamayor-Polo, L. et al. Immunologic evaluation and genetic defects of apoptosis in patients with autoimmune lymphoproliferative syndrome (ALPS). Crit. Rev. Clin. Lab. Sci. 58, 253–274 (2021).
4. Bleesing, J. J., Nagaraj, C. B. & Zhang, K. Autoimmune Lymphoproliferative Syndrome. in GeneReviews® (eds. Adam, M. P. et al.) (University of Washington, Seattle, Seattle (WA), 1993).
Para mayor información o consultas:
Sección: Citometría de Flujo
Téc. María Victoria Rodríguez
Dr. Mariano M. Gonzalez
Tel: 0341-4861600. Int: 259
Sección: Biología Molecular
Lic. Alan Gomez
Lic. Analia Seravalle
Tel: 0341-4861600. Int: 225 / 242