

El Síndrome de Gilbert (SG) es una condición genética benigna caracterizada por episodios intermitentes de hiperbilirrubinemia no conjugada, generalmente sin daño hepático. Se presenta en aproximadamente el 5–10 % de la población general, y suele detectarse durante la adolescencia o adultez temprana, muchas veces de manera incidental al realizar análisis de rutina. Los síntomas, cuando aparecen, suelen desencadenarse por ayuno, estrés, enfermedad, ejercicio físico o consumo de ciertos medicamentos [1].
Genética del síndrome: el gen UGT1A1
El SG está asociado con variantes en el gen UGT1A1, que codifica la enzima UDP-glucuronosiltransferasa 1A1, responsable de la conjugación de bilirrubina en el hígado. La variante más común, particularmente en poblaciones caucásicas, es la inserción de dos nucleótidos TA en la región promotora (variante UGT1A1*28), que reduce significativamente la expresión génica [1,2].
En poblaciones asiáticas, son frecuentes otras variantes como UGT1A1*6 (c.211G>A, p.Gly71Arg) y UGT1A1*7, que son variantes missense en la región codificante del gen y también afectan la función enzimática [3].
La herencia del SG suele ser autosómica recesiva, aunque hay evidencia de expresión variable en heterocigotas, y en ciertos casos puede observarse un patrón autosómico dominante con penetrancia incompleta [2].
Importancia clínica del test genético
La determinación de variantes en el gen UGT1A1 tiene múltiples aplicaciones clínicas:
• Confirmación diagnóstica del SG en pacientes con hiperbilirrubinemia no conjugada leve persistente [1].
• Diferenciación con otros trastornos más graves como los síndromes de Crigler–Najjar tipo I y II [2].
• Aplicaciones farmacogenómicas: ciertas variantes de UGT1A1 están asociadas a toxicidad aumentada por fármacos como irinotecán (antineoplásico), atazanavir (antirretroviral), y otros metabolizados por glucuronidación [4,5].
• Valor en asesoramiento genético, especialmente en estudios familiares y en planificación terapéutica individualizada [3].
Prestaciones disponibles en Cibic Laboratorios:
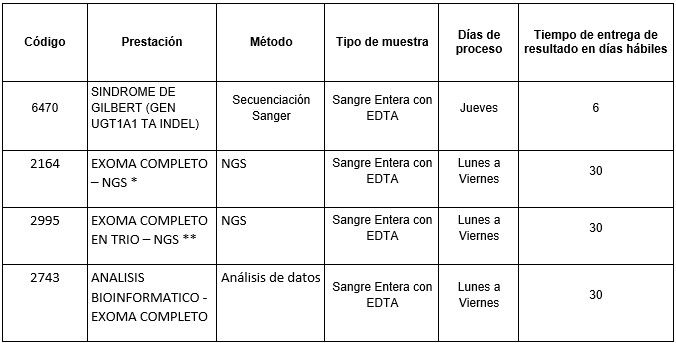
* Exoma completo dirigido a patología especifica con diagnóstico presuntivo (HPO), con o sin análisis del genoma mitocondrial.
**Exoma completo trío incluye a los progenitores del probando con el fin de aumentar el rendimiento diagnóstico de la prueba
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Referencias
1. Grant LM, Faust TW, Thoguluva Chandrasekar V, et al. Gilbert Syndrome. [Updated 2024 Oct 5]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470200/.
2. https://omim.org/entry/143500.
3. De Silva AP, Nuwanshika N, Niriella MA, de Silva HJ. Gilbert’s syndrome: The good, the bad and the ugly. World J Hepatol. 2025 Feb 27;17(2):98503. doi: 10.4254/wjh.v17.i2.98503. PMID: 40027563; PMCID: PMC11866151.
4. Bosma PJ. Inherited disorders of bilirubin metabolism. J Hepatol. 2003 Jan;38(1):107-17. doi: 10.1016/s0168-8278(02)00359-8. PMID: 12480568.
5. Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, Karrison T, Janisch L, Ramírez J, Rudin CM, Vokes EE, Ratain MJ. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004 Apr 15;22(8):1382-8. doi: 10.1200/JCO.2004.07.173. Epub 2004 Mar 8. PMID: 15007088.
6. Incorporación del estudio Exoma Completo a nuestro portfolio de genómica
www.cibic.com.ar/cibic/incorporacion-estudio-exoma-completo-portfolio-genomica
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Alan Gomez.
Tel +54 (341) 4861600 (Int. 225)
Sección: Biología Molecular
Lic. Analía Seravalle.
Tel +54 (341) 4861600 (Int 242