
{kind=link}
El síndrome MELAS (Encefalomiopatía mitocondrial, acidosis láctica y episodios semejantes a apoplejías) constituye una enfermedad neurodegenerativa multisistémica progresiva caracterizada por episodios agudos neurológicos comparables a la apoplejía, asociados a la hiperlactatemia y la miopatía mitocondrial. A menudo se producen en pacientes con síntomas crónicos como debilidad muscular, sordera, diabetes, baja estatura, miocardiopatía, retraso en el desarrollo, pérdidas de memoria o trastornos de atención. Se desconoce la prevalencia exacta de la enfermedad. Los síntomas clínicos comienzan generalmente durante la infancia o la edad adulta temprana.
![]() MELAS es causada por mutaciones a nivel del ADN mitocondrial (Mitochondrial DNA, mtDNA) y es transmitida por herencia materna. El diagnóstico de MELAS se basa en la combinación de hallazgos clínicos y estudios genéticos. Mutaciones en el gen MT-TL1 localizado a nivel del ADN mitochondrial (mtDNA) que codifica para tRNA Leu (UUA/UUG) se hallan asociadas a MELA. La mutación m.3243A>G es la más comúnmente asociada a MELAS (80% de los casos) (1).
MELAS es causada por mutaciones a nivel del ADN mitocondrial (Mitochondrial DNA, mtDNA) y es transmitida por herencia materna. El diagnóstico de MELAS se basa en la combinación de hallazgos clínicos y estudios genéticos. Mutaciones en el gen MT-TL1 localizado a nivel del ADN mitochondrial (mtDNA) que codifica para tRNA Leu (UUA/UUG) se hallan asociadas a MELA. La mutación m.3243A>G es la más comúnmente asociada a MELAS (80% de los casos) (1).
Este tipo de mutaciones pueden ser detectadas en mtDNA de leucocitos, generalmente en aquellos individuos que presenten el diagnóstico para MELAS clásico. Sin embargo, debido a la presencia de heteroplasmia, es posible que la mutación a ser identificada esté presente en diferentes distribuciones en función al tejido seleccionado para el estudio. Debido a esto, se recomienda ante un resultado negativo en mtDNA de leucocitos, realizar la prueba en otro tipo de tejidos (fibroblastos cultivados, folículo piloso, sedimento urinario y en última instancia, biopsia de músculo esquelético) (2,3).
Determinación disponible en Cibic:
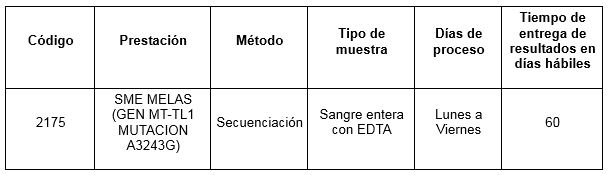
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la intranet.
Bibliografía
– Goto Y y cols. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature.1990;348:651–3.
– McDonnell MT y cols. Noninvasive diagnosis of the 3243A > G mitochondrial DNA mutation using urinary epithelial cells. Eur J Hum Genet. 2004;12:778–81.
– Shanske S y cols. Varying loads of the mitochondrial DNA A3243G mutation in different tissues: implications for diagnosis. Am J Med Genet A. 2004;130A:134–7.
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Analía Seravalle Tel: 0341 4499444 Int: 242
Lic. Guadalupe Méjico Tel: 0341 4499444 Int: 243
Dra. María Florencia Gosso Tel: 0341 4499444 Int: 258