

La Ataxia de Friedrich (FRDA) es un trastorno neurodegenerativo hereditario que se caracteriza clásicamente por una ataxia progresiva de la marcha, disartria, disfagia, disfunción oculomotora, pérdida de los reflejos tendinosos profundos, signos de afectación del tracto piramidal, escoliosis, y en algunos casos, miocardiopatía, diabetes mellitus, pérdida visual y audición defectuosa. La presentación clásica de la FRDA se inicia en la niñez o adolescencia.
Se estima que la prevalencia de la FRDA en la población caucásica es de 1/20.000 a 1/50.000.
La FRDA está causada mayormente (98% de los casos) por una expansión en homocigosis del triplete GAA situado en el intrón 1 del gen FXN (9q21.11) que codifica la frataxina. Actualmente se desconoce la función de esta proteína, pero la teoría más aceptada es que juega un papel en la biogénesis de los núcleos hierro-azufre. Una deficiencia en esta proteína conduce al daño progresivo en el sistema nervioso central y periférico visto en la FRDA.
En personas que presentan FRDA, el segmento GAA se repite 66 a más de 1,000 veces. La longitud de la repetición del trinucleótido GAA parece estar relacionada con la edad a la que aparecen los síntomas, qué tan graves son y qué tan rápido progresan. Las personas con segmentos GAA repetidos menos de 300 veces tienden a tener una aparición más tardía de síntomas (después de los 25 años) que aquellas con repeticiones más grandes.
Por otro lado, pueden presentarse casos de FRDA en individuos heterocigotos compuestos (2-5%), los cuales poseen una expansión del trinucleótido GAA en un alelo y una mutación puntual en el otro alelo.
Tests genéticos
La FRDA se hereda de forma autosómica recesiva, siendo posible el consejo genético por parte de un especialista. Los test genéticos moleculares son importantes en este contexto ya que permiten la identificación de mutaciones en el gen FXN, confirmando el diagnóstico. Mediante la técnica conocida como triplet repeat primed PCR (TP-PCR) es posible determinar el número de repeticiones de GAA, mientras que la secuenciación completa del gen FXN permite la identificación de mutaciones puntuales.
Diagnóstico diferencial
El diagnóstico diferencial incluye Charcot-Marie-Tooth tipos 1 y 2, ataxia con deficiencia de vitamina E, ataxia-apraxia oculomotora tipos 1 y 2 y otras ataxias de aparición temprana.
No existe cura para la FRDA y su manejo es multidisciplinar. Su pronóstico ha mejorado, pero la calidad de vida aún está significativamente afectada. La esperanza de vida es de unos 40 años de media, dependiendo de la edad de aparición y la presencia de diabetes y miocardiopatía.
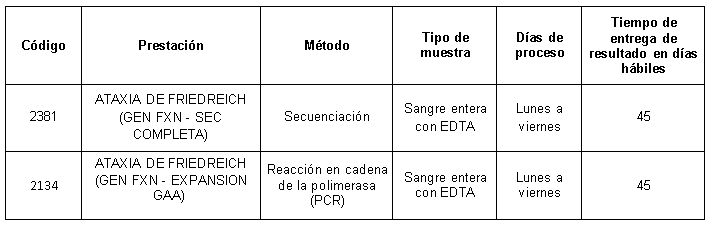
Prestaciones disponibles en Cibic:
Referencias:
– Orphanet. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=ES&Expert=580
– https://ghr.nlm.nih.gov/condition/friedreich-ataxia#inheritance
– Rao, Vamshi & Didonato, Christine & Larsen, Paul. (2018). Friedreich’s Ataxia: Clinical Presentation of a Compound Heterozygote Child with a Rare Nonsense Mutation and Comparison with Previously Published Cases. Case Reports in Neurological Medicine. 2018. 1-5. 10.1155/2018/8587203.
Para mayor información o consultas:
Sección: Biología Molecular
 Lic. Alan Gomez.
Lic. Alan Gomez.
Tel 0341-4722424. Interno: 243/225
Sección: Análisis Especiales
 Lic. Analía Seravalle.
Lic. Analía Seravalle.
Tel 0341-4722424. Interno: 242.