

El Síndrome de Lynch (SL), también llamado cáncer colorrectal (CCR) hereditario no polipósico (CCRHNP), es un síndrome genético, heterogéneo, con patrón de herencia autosómico dominante y penetrancia incompleta. Este síndrome predispone al desarrollo de tumores en diferentes localizaciones, principalmente en el colon (y recto) y útero (endometrio), y con menos frecuencia en el ovario, intestino delgado, estómago, las vías urinarias, vía biliar, páncreas, próstata, entre otros.
Su prevalencia en la población general se estima en 1:440 y es causa del 1 al 3 % del total de casos de cáncer colorrectal y del 0,8 al 1,4% de casos de cáncer de endometrio
Se atribuye a mutaciones heredadas de forma autosómica dominante en genes implicados en la reparación de errores que ocurren durante la replicación del DNA (MMR, del inglés mismatch repair) previo a la división celular. En la mayoría de los casos se debe a mutaciones en los genes MLH1, MSH2, MSH6 y PMS2, así como a deleciones germinales del gen EPCAM, el cual no es un gen MMR pero causa la inactivación del gen MSH2
Su inactivación, debida a mutaciones puntuales, pequeñas deleciones e inserciones, o grandes rearreglos génicos (deleciones e inserciones más largas) origina inestabilidad genética, provocando indirectamente la aparición del tumor por acumulación de mutaciones en otros genes.
Aproximadamente el 90% de casos de SL se deben a variantes patogénicas germinales en los genes MLH1 y MSH2, seguido de MSH6 en el 7%-10% de los casos y PMS2 en menos de 5% de los casos; mientras que las deleciones en el gen EPCAM son la causa en ~1% de los casos.
De acuerdo al tipo de variante patogénica identificada en los genes MMR, se han identificado distintos fenotipos del SL.
Las variantes patogénicas en el gen MLH1 son más frecuentes en pacientes con CCR mientras que las variantes en el gen MSH2 son más frecuentes en pacientes con neoplasias extracolónicas y en el síndrome de Muir-Torre.
Las variantes patogénicas en el gen MSH6 se relacionan principalmente con cáncer de endometrio y de presentación más tardía comparado con pacientes con variantes en los otros genes MMR. Se ha sugerido que las variantes patogénicas en el gen PMS2 conllevan a presentar un fenotipo intermedio, de menor riesgo a desarrollar cáncer, así como ser de presentación tardía.
Las deleciones germinales en el gen EPCAM resultan en el silenciamiento del gen MSH2, asociándose a un riesgo incrementado a desarrollar CCR e incluso se ha propuesto que el riesgo a desarrollar neoplasias extracolónicas depende de la extensión de la deleción.
Con edades tempranas de aparición entre los 40 y 50 años, los portadores de una mutación tienen un riesgo de desarrollar un cáncer colorrectal a lo largo de la vida de alrededor del 60-70%. El riesgo de desarrollar un nuevo tumor primario en aquellos pacientes que ya han presentado uno es del 15% durante un período de 10 años, y del 40% en un período de 20 años, si no se ha resecado todo el colon.
Pruebas genéticas.
Cuando la historia personal o familiar indica la posibilidad de un síndrome de cáncer hereditario, el análisis genético es de gran interés y utilidad clínica. Es importante resaltar que la indicación de análisis genético debe provenir de un equipo médico especializado que evaluará cada caso en particular. La detección de una mutación tiene implicancias importantes en el portador y en sus parientes, puesto que cada familiar de primer grado de un portador, padres, hijos o hermanos, tiene la probabilidad de un 50% de ser portador.
Los estudios genéticos moleculares de los genes MMR permiten identificar una variante patogénica cuando los hallazgos clínicos y del tumor son consistentes con SL.
El diagnóstico definitivo del SL se establece mediante la identificación de alguna variante patogénica en los genes MMR o en el gen EPCAM, mediante secuenciación o análisis de deleciones/duplicaciones.
La secuenciación y screening de variantes en el número de copias (CNVs) se lleva a cabo a través de un panel multigenético que incluye los genes MLH1, MSH2, MSH6, PMS2 y EPCAM (solo screening CNVs) el cuál se realiza mediante la metodología de Secuenciación Masiva (NGS, Next Generation Sequencing).
Aquellas variantes puntuales o pequeñas indels (inserciones/deleciones), identificadas en el caso índice luego pueden ser estudiadas en los familiares de este mediante secuenciación tipo Sanger. Las variantes identificadas por screening de CNVs deben ser confirmadas por MLPA (Multiplex Ligation-dependent Probe Amplification)
El análisis de deleciones/duplicaciones se lleva a cabo mediante MLPA, que es la técnica gold standard para el estudio de este tipo de alteraciones.
Héritas, empresa especializada en análisis genéticos asociada a Cibic Laboratorios cuenta con un Servicio de Asesoramiento Genético que, a la vista de los antecedentes familiares del paciente, evalúa la posibilidad de proceder a un estudio con el fin de determinar si posee alguna alteración genética que suponga una predisposición a padecer síndrome de Lynch.
La identificación de los portadores de mutaciones permite aplicar medidas de seguimiento, detección precoz y prevención.
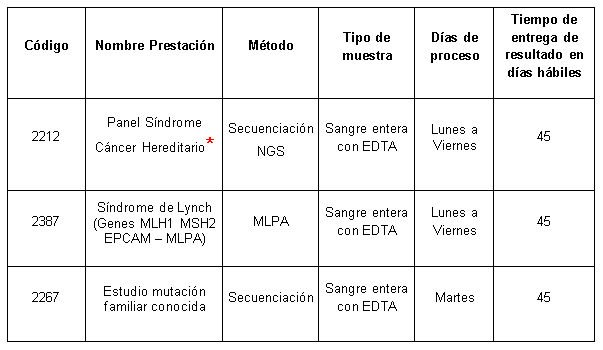
Determinaciones disponibles en Cibic:
* https://heritas.com.ar/genomica-clinica/cancer-hereditario/
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre la práctica consulte al manual de prestaciones y a la extranet.
Referencias
1 María del Carmen Castro-Mujica, Claudia Barletta-Carrillo. Síndrome de Lynch: aspectos genéticos, clínicos y diagnósticos. Rev Gastroenterol Peru. 2018;38(3):265-79
2 McKusick VA. Lynch Syndrome. Baltimore, MD: OMIM; c 1966-2018. Disponible en: http://omim.org/entry/120435
3 Lynch HT, Lynch JF, Shaw TG. Hereditary gastrointestinal cancer syndromes. Gastrointest Cancer Res GCR. 2011;4(4 Suppl 1):S9-17.
4 Kohlmann W y cols.. SourceGeneReviews® [Internet]. Seattle (WA): University of Washington; c1993-2018. 2004 Feb 5 [updated 2018 Apr 12].
5 Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919-32.
6 Steinke V y cols. Hereditary nonpolyposis colorectal cancer (HNPCC)/ Lynch syndrome. Dtsch Arztebl Int 110(3):32-38, 2013.
7 Rasmussen LJ y cols. Pathological assessment of mismatch repair gene variants in Lynch Syndrome: past, present, and future. Hum Mutat 33(12):1617-1625, 2012.
8 Síndrome de cáncer hereditario. https://www.cibic.com.ar/home/sindrome-cancer-hereditario/
Para mayor información o consultas:
 Sección: Biología Molecular
Sección: Biología Molecular
Lic. Alan Gomez.
Tel 0341-4722424. Interno: 243/225
 Sección: Análisis Especiales
Sección: Análisis Especiales
Lic. Analía Seravalle.
Tel 0341-4722424. Interno: 242.