

El mieloma múltiple (MM) es el segundo cáncer hematológico más común en adultos. Es una neoplasia resultante de la proliferación de células plasmáticas inmunosecretoras.
Esta patología tiene una frecuencia del 13% de los procesos oncohematológicos, con una incidencia de 4 casos/100.000 habitantes/año. Se presenta principalmente en adultos de entre 50 a 80 años, con mayor prevalencia en el sexo masculino.
Al momento del diagnóstico, entre el 15 y 30% de los pacientes son asintomáticos.
Cuando son sintomáticos pueden presentar:
• debilidad y fatiga
• dolor de huesos
• proteína monoclonal demostrable en suero u orina
• inmunoglobulina normal disminuida
• anemia
• hiperuricemia
• plasmocitosis de la médula ósea
• lesiones óseas líticas
• función renal anormal
• aumento en el número de células plasmáticas (1)
En el 60-70% de los pacientes con MM se observa un cariotipo aparentemente normal. Aunque muchas alteraciones genéticas son susceptibles de detección por citogenética convencional, su detección se ve dificultada por el bajo índice proliferativo de células plasmáticas y, en consecuencia, el número limitado de metafases.
Con la incorporación de estudios de hibridación in situ con fluorescencia en interfase (FISH) se pueden detectar alteraciones genéticas en más del 80% de los casos, incluso alteraciones crípticas no detectadas mediante citogenética convencional.
Los estudios de citogenética y genética molecular han emergido como factores pronósticos relevantes, capaces de identificar pacientes con diferentes características clínicas y respuesta a la terapia. Pueden distinguirse inicialmente anomalías primarias, directamente relacionadas con la patogenia de la enfermedad y eventos secundarios, asociados con la progresión de la enfermedad.
Las alteraciones primarias se pueden subdividir en dos categorías citogenéticas:
1) Hiperdiploides (45% de los casos): se caracterizan por la presencia de trisomías de los cromosomas impares y una baja frecuencia de translocaciones que involucran el locus 14q32 (cadena pesada de inmunoglobulina IGH). En general, muestran un comportamiento clínico más favorable.
2) No hiperdiploides (40% de los casos, abarca hipodiploide, pseudodiploide y casi tetraploide MM): se caracterizan por una alta frecuencia de translocaciones que involucran el locus 14q32 (IGH).
Las categorías de ploidía son estables en el tiempo, y rara vez cambia en la progresión de la enfermedad. En un pequeño subgrupo de pacientes (10 %), se pueden observar translocaciones de IGH e hiperdiploidia. Esta asociación es considerada desfavorable.
Además, existe un subgrupo de casos (15%) que presentan otro tipo de alteraciones y curso clínico variable.
Dentro de las translocaciones que involucran el locus 14q32 (IGH) podemos encontrar:
– Translocación t(11;14)(q13;q32): es la translocación más frecuente de este grupo y ocurre en aproximadamente 15-20% de los pacientes con MM recién diagnosticados. Asociado con una evolución clínica favorable; la enfermedad puede ser heterogénea y su efecto global sobre el pronóstico se considera neutral. Una evaluación reciente de su impacto en el resultado del trasplante autólogo de células hematopoyéticas concluye que la t(11;14) tiene un peor resultado que los pacientes con cariotipo y estudios de FISH normales, pero mejor que los pacientes con marcadores de alto riesgo.
– Translocación t(4;14)(p16.3;q32): ocurre en aproximadamente el 15% de los pacientes. Es una translocación críptica, involucra dos genes codificadores de proteínas, MMSET y FGFR3. Se asocia al uso de cadena pesada de IgA y una prevalencia muy alta de deleción/monosomía del cromosoma 13. Se consideran de riesgo intermedio en algunos estudios y de alto riesgo en otros.
– Translocaciones menos frecuentes: t(14;16)(q32;q23) (5%), t(14;20)(q32;q11) (2%) y t(8;14)(q24.3;q32) (<1%) que involucran genes c-MAF, MAFB y MAFA, respectivamente. Asociados con mayor frecuencia a deleción del cromosoma 13 y un resultado clínico más agresivo.
La presencia de eventos genéticos secundarios refleja la progresión del tumor. Se han descrito alteraciones secundarias, siendo las más frecuentes:
• Deleción/ monosomía del cromosoma 13: se detectan alteraciones del cromosoma 13 en el 50% de los casos, el 85% monosomías, mientras que el 15% restante son deleciones intersticiales. Esta alteración se asoció primero con un pronóstico desfavorable y una supervivencia corta, pero cada vez hay más pruebas de que su relevancia pronóstica puede estar relacionada con su asociación con translocaciones de IGH, particularmente t(4;14) (90% de los casos) considerándose como un marcador de MM no hiperdiploide.
• Deleción cromosoma 17p13: se considera el factor pronóstico citogenético molecular más importante en pacientes con MM. La proteína supresora de tumor TP53 tiene un papel importante en la apoptosis, senescencia o detención del ciclo celular, mientras que la deleción o las mutaciones de TP53 pueden predisponer a las células al daño del ADN o permitir la supervivencia celular. Ocurre tardíamente en MM, se informa en aproximadamente el 10% de los casos. Su presencia predice una supervivencia más corta, enfermedad más agresiva, mayor prevalencia de enfermedad extramedular, hipercalcemia, corta duración de la respuesta post-alta a la terapia y compromiso del sistema nervioso central.
• Alteraciones del cromosoma 1 (deleciones 1p y ganancias/ amplificaciones 1q): encontrado en hasta el 45% de MM. Esta alteración introduce un mayor nivel de inestabilidad genética en células de mieloma y sugieren la amplificación 1q como un posible marcador subrogado de tumores clonalmente más avanzados. Se observó una supervivencia significativamente más corta de pacientes que presentan esta alteración comparada con aquellos que no la presentan. Además, se encontró mayor frecuencia de ganancia 1q en pacientes con recaída, probablemente asociado a la resistencia al tratamiento.
• Los reordenamientos de MYC (8q24): implica alteraciones complejas, no recíprocas, duplicaciones, amplificaciones que puede ser mediado por eventos secundarios que no involucran mecanismos de recombinación específicos de células B, y a veces no involucran el loci de inmunoglobulina. Estas alteraciones ocurren en el 15% de los pacientes recién diagnosticados con MM, pero se observan en el 45% de los casos con enfermedad avanzada y en casi el 90% de las líneas celulares de mieloma humano, mostrando una prevalencia similar en los casos hiperdiploide y no hiperdiploide. Las translocaciones que involucran los loci MYC e IGH son frecuentemente observados como un evento tardío durante la progresión del tumor cuando las enfermedades son cada vez más proliferativas y menos dependiente del estroma. La activación de MYC también se detectó en pacientes con MGUS, lo que sugiere que podría ser un evento genético temprano en la patogenia del MM.
• Otras alteraciones: algunos pacientes presentan un curso clínico heterogéneo. Un bajo porcentaje tienden a tener deleción del brazo corto del cromosoma 12, esto ocurre en alrededor del 8% de los pacientes con MM y en el 24% de los casos con leucemia de células plasmáticas. El tamaño de la deleción es variable, y tiende a aparecer en un estadio avanzado de la enfermedad, que representa un cambio secundario asociado con progresión de la enfermedad.
Otra variable a considerar es la presencia de cariotipos complejos que son consecuencia de la acumulación de cambios genéticos secuenciales que aparecen durante el desarrollo de clones tumorales y están asociados con la progresión de la enfermedad. (2)
Las anomalías genéticas se pueden clasificar como:

En Cibic, para el estudio y seguimiento de mieloma múltiple, contamos con análisis de cariotipo y estudio de FISH (Hibridación in situ fluorescente).
Las sondas correspondientes a dicha patología pueden ser solicitadas de manera individual, o bien, mediante paneles:
Panel MM1:
1p32(CDKN2C)/1q21(CKS1B)
13q14(RB1)
14q32(IGH)
17p13(TP53)
Panel MM2:
t(4;14)(FGFR3;IGH)
t(11;14)(CCND1;IGH)
t(14;16)(IGH;MAF)
t(14,20)(IGH;MAFB)
El Panel MM2 se realizará en base al resultado correspondiente a 14q32(IGH).
Prestaciones disponibles en Cibic Laboratorios:
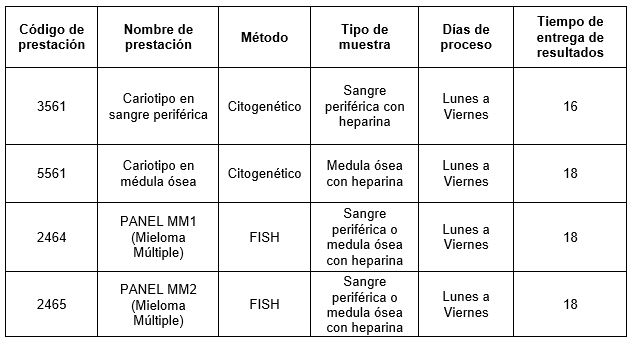
Para solicitar las sondas individuales:
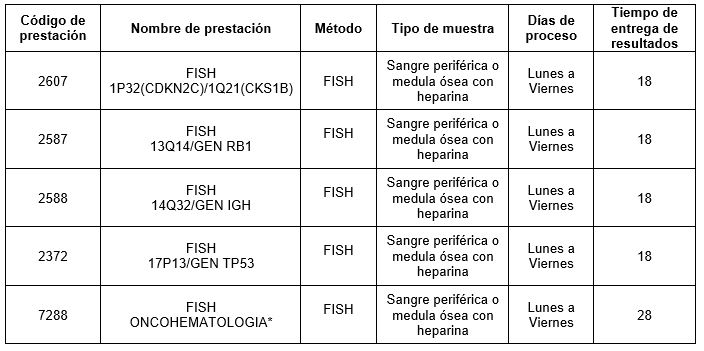
*indicar sonda solicitada:
• t(4;14)(p16.3;q32.3): IGH/FGFR3
• t(11;14)(q13;q32): CCND1(BCL1)/IGH
• t(14;16)(q32.3;q22): IGH/MAF
• t(14;20)(q32;q12): IGH-MAFB
• 8q24: MYC
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Referencias
1. Flavia Stella, Estela Pedrazzini, Mara Agazzoni, Oscar Ballester e Irma Slavutsky (2015): Alteraciones citogenéticas en el mieloma múltiple: importancia pronóstica y elección de la terapia de primera línea, Investigación del cáncer, DOI: 10.3109/07357907.2015.1080833
2. Marilyn S. Arsham, Margaret J. Barch, Helen J. Lawce. The AGT Cytogenetics Laboratory Manual. Pág. 515 Cuarta edición- 2017.
Para mayor información o consultas:
Sección: Citogenética

Bioq. Sibila Bertalot

Lic. Veronica Vanrell
 Téc. Victorina Carbone
Téc. Victorina Carbone
Tel: 0341-4861600. Int: 283