

La anemia de Fanconi (FA, por sus siglas en inglés) es una enfermedad genética y fenotípicamente heterogénea, caracterizada por fallo medular, malformaciones congénitas, disfunción endocrina e incremento en la predisposición a desarrollar de distintos tipos de cánceres, entre otras alteraciones. En más de un 98%, la FA se hereda con un patrón de herencia autosómico recesivo. En un porcentaje menor de casos se halla ligada al cromosoma X (1-2%) (1, 2).
La FA es una enfermedad rara que puede resultar difícil de diagnosticar debido a la amplia variedad de síntomas. Anormalidades congénitas como microcefalia, microftalmia, anomalías de radio o pulgares, trastornos del crecimiento, entre otros son síntomas que pueden orientar sobre la presencia de la enfermedad. Hematológicamente los signos más comunes que pueden presentar son anemia aplásica, síndrome mielodisplásico (SMD), leucemia mieloide aguda y una o más citopenias de cualquier linaje. Tanto el SMD como la LAM son primarios y de presentación a temprana edad. Además estos individuos tienen una elevada predisposición para desarrollar tumores sólidos. Historia familiar de cánceres y/o antecedentes de hipersensibilidad al tratamiento por radiación o quimioterapia son signos de sospecha de la enfermedad (1, 2).
Hasta el momento, se han identificado mutaciones en 21 genes diferentes. De esta manera se han definido 21 subtipos o grupos de complementación. La mayoría de los pacientes (60-70 %) que desarrollan esta enfermedad pertenecen al grupo FA-A. Le siguen en porcentaje los grupos FA-C y FA-G, con 16% y 10%, respectivamente (3)
Se ha confirmado que las proteínas codificadas por estos 21 genes forman parte de la vía de señalizaciones denominada FA-BRCA. Éstas tienen como función principal participar de la reparación de los entrecruzamientos intercadena del ADN que generan rupturas y aberraciones cromosómicas (3). La deficiencia en la vía de señalización encargada de la reparación de los entrecruzamientos entre las cadenas de ADN se manifiesta como fragilidad cromosómica espontánea y además en una elevada sensibilidad a la exposición a agentes clastogénicos (4).
Esta hipersensibilidad es la base de las pruebas diagnósticas básicas. Agentes clastogénicos como el diepoxibutano (DEB), mitomicina C (MMC) o cis-platino inducen la generación de estos puentes entre las hebras de ADN. De esta manera los pacientes con FA muestran un incremento en la formación de estructuras cromosómicas radiales, roturas, gaps, e intercambios de cromátides ante la exposición a estos agentes clastogénicos. La incapacidad de las células para responder adecuadamente a un daño de este tipo determina que, los individuos con FA presente de manera espontánea, en sus células una alta frecuencia de aberraciones cromosómicas (5, 6).
La prueba de sensibilidad a DEB se considera como estándar de oro ya que permite analizar aberraciones cromosómicas espontáneas e inducidas por este agente. A partir de cultivos de linfocitos de sangre periférica expuestos a DEB, se analiza la fragilidad cromosómica identificando rupturas cromatídicas y cromosómicas, presencia de fragmentos céntricos y acéntricos, figuras radiales, translocaciones, anillos, etc.
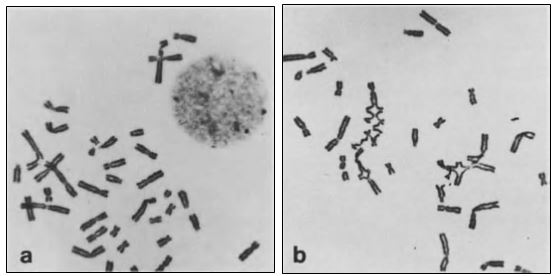
Figuras: Metafases parciales obtenidas a partir de cultivos de linfocitos de sangre periférica de un individuo con FA.
a) Se muestra la aberración espontánea.
b) Luego de la exposición de las células a 0,1 µg/ml DEB. Se observa el incremento de las anomalías cromosómicas pos-tratamiento con el agente clastogénico.
Fuente: Auerbach et al. (1981)
El número de células que se analizan varía entre 50 y 100 por individuo y por tratamiento. Así mismo se analiza en paralelo una muestra de un individuo sano, ya que las condiciones del cultivo o el agente al que se exponen las células pueden variar. Las diferencias de la frecuencia de aberraciones cromosómica de un paciente con FA son diez veces más que las presentes en un individuo sin FA (5, 6).
Algunos pacientes con FA tienen células normales (mosaicos), producto generalmente de reversión de la mutación o por conversión génica (1). En individuos con mosaico para FA estas pruebas se dificultan debido a la disminución en la frecuencia de aberraciones cromosómicas por la presencia de estas células, hecho que puede dar lugar a falsos negativos. Es común que los pacientes reciban varias transfusiones por lo que se recomienda realizar el estudio después de por lo menos tres meses de la última transfusión. Una alternativa a esta situación podría ser realizar el test en cultivo de fibroblastos (5, 6).
En el área de la biología molecular se cuenta con metodologías como la MLPA y la secuenciación completa para el gen FANCA. Mutaciones en FANCA pueden producir una proteína FANCA anormal o mutaciones asociadas a la ausencia total de proteína. La edad de inicio de la anemia y la incidencia de leucemia es más temprana en aquellos pacientes con mutaciones asociadas a la ausencia total de proteína (7).
Ante la sospecha clínica de FA, estos ensayos de hipersensibilidad utilizando agentes clastogénicos son una herramienta útil en el diagnóstico temprano de la enfermedad. Lo que permite excluir otras enfermedades y previene un manejo inadecuado del paciente, e indicar el asesoramiento genético para las familias en riego.
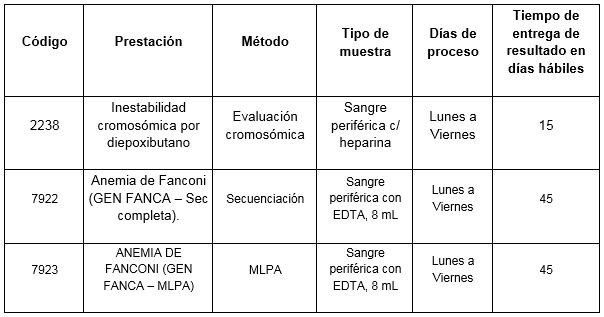
Prestaciones disponibles en Cibic:
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Bibliografía
1. Fanconi Anemia: Guidelines for Diagnosis and Management, Fourth Edition, 2014
2. Sociedad Argentina de Hematología. Guías de Diagnóstico y tratamiento. 2015
3. Mamrak NE., Shimamura A., Howlett NG. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. Oct 13, 2016
4. Kupfer GM. Fanconi anemia: a signal transduction and DNA repair pathway. Yale J Biol Med. 86: 491-497, 2013
5. Auerbach AD. Diagnosis of Fanconi anemia by diepoxybutane analysis. Curr Protoc Hum Genet. Apr 1;85:8.7.1-17, 2015
6. Schroeder-Kurth T. M., Auerbach A. D. Fanconi anemia: clinical, cytogenetic, and experimental aspects. G.Obe (eds.). Springer-Verlag Berlin Heidelberg 1989.
7. Faivre L et al. Association of complementation group and mutation type with clinical outcome in Fanconi anemia. European Fanconi Anemia Research Group. Blood. 2000; 96:4064-70.
Para mayor información o consultas:
 Sección: Citogenética.
Sección: Citogenética.
Bioq. Carlos Zumoffen.
Tel: 0341-4499444 interno 283.

Sección: Biología Molecular
Bioq. María Florencia Gosso.
Tel: 0341-4499444. Interno: 258