

Las distrofinopatías cubren un espectro de enfermedad muscular que varía de leve a grave ligada al cromosoma X, debido a mutaciones en el gen DMD (MIM:*300377) que codifica una proteína llamada distrofina que se expresa en las células musculares. En estas se incluyen la distrofia muscular de Duchenne (DMD, MIM:#310200), la distrofia muscular de Becker (DMB, MIM#300376) y la miocardiopatía dilatada asociada a DMD (MCD, MIM#302045) (1).
El extremo leve del espectro incluye los fenotipos de aumento asintomático en la concentración sérica de creatina fosfoquinasa (CK) y calambres musculares con mioglobinuria.
El extremo severo del espectro incluye enfermedades musculares progresivas que se clasifican como distrofia muscular de Duchenne / Becker cuando el músculo esquelético se ve afectado principalmente y como miocardiopatía dilatada asociada a DMD (MCD) cuando se ve afectado principalmente el corazón (1).
La DMD, con una incidencia de 1 por cada 3.300 nacimientos de varones (1), suele presentarse en la primera infancia con retrasos en los hitos motores, incluyendo retrasos en el caminar de forma independiente y en ponerse de pie desde una posición supina. La debilidad proximal causa una marcha de pato y dificultad para subir escaleras, correr, saltar y ponerse de pie en cuclillas. La DMD progresa rápidamente, llevando a requerir el uso de sillas de ruedas. La cardiomiopatía se presenta en casi todos los individuos con DMD después de los 18 años.
La DMB se caracteriza por la debilidad muscular esquelética de aparición tardía, la incidencia varía entre 1 por cada 18.000 – 31.000 nacimientos de varones (2). Gracias a las mejores técnicas de diagnóstico, se ha reconocido que el extremo leve del espectro incluye a los hombres con inicio de síntomas después de los 30 años de edad que permanecen ambulantes incluso hasta los 60 años. A pesar de la afectación más leve del músculo esquelético, la insuficiencia cardíaca por DMB es una causa común de morbilidad (1).
La MCD se caracteriza por la dilatación del ventrículo izquierdo y la insuficiencia cardíaca congestiva. Las mujeres heterocigotas para una variante patógena en DMD tienen un mayor riesgo de padecer MCD (1).
El gen DMD, localizado en el cromosoma X (Xp21.2) es el gen humano más grande conocido, y codifica una proteína llamada distrofina, que se expresa en las células musculares. Las mutaciones responsables de DMD conllevan una ausencia total de distrofina, mientras que las que subyacen a la DMB conducen a una cantidad y/o calidad anormal de la proteína (3).
En ambos casos es frecuente encontrar deleciones del gen completo o de varios exones (60-65%) y duplicaciones de uno o más exones (5-10%). En menor frecuencia, pueden hallarse mutaciones puntuales (20-35%). Es en base a estos datos que se plantea el algoritmo diagnostico a seguir ante la sospecha de una distrofinopatía (ver Figura 1), el cual se recomienda comenzar evaluando duplicaciones y/o deleciones en el gen DMD (4), preferentemente mediante MLPA ya que el CMA array no es capaz de detectar todas las deleciones/duplicaciones dentro del gen (1).
De no hallarse ninguna variante patogénica mediante el análisis de deleciones/duplicaciones, se debe continuar evaluando mutaciones puntuales mediante secuenciación completa del gen DMD, en este punto se puede considerar un panel multigenético (mediante secuenciación NGS) que incluya además genes asociados a distrofia muscular de cinturas y distrofia muscular de Emery-Dreifuss, principalmente en individuos con presentaciones clínicas menos severas (1).
Es importante aclarar que en los paneles multigenéticos que incluyen el gen DMD también se realiza un screening de CNVs (variante del número de copias) de dicho gen, mediante el cual se pueden detectar deleciones y/o duplicaciones, aunque tales hallazgos luego deben confirmarse por MLPA.
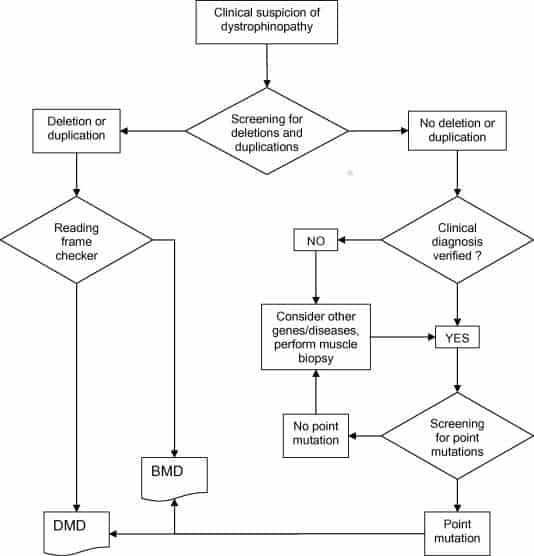
Figura 1. Algoritmo diagnostico propuesto para distrofinopatías, tomado de Abbs S. et al. (2010).
En el diagnóstico diferencial se deben también considerar la atrofia muscular espinal (AME) y el Síndrome de Barth (1).
Las distrofinopatías se heredan de forma ligada al X. Las mujeres portadoras tienen un riesgo del 50% de transmitir la mutación a su descendencia. Los hijos varones que hereden la mutación estarán afectados; las hijas que hereden la mutación serán portadoras, y poseen un elevado riesgo de desarrollar cardiomiopatía dilatada asociada a DMD (3).
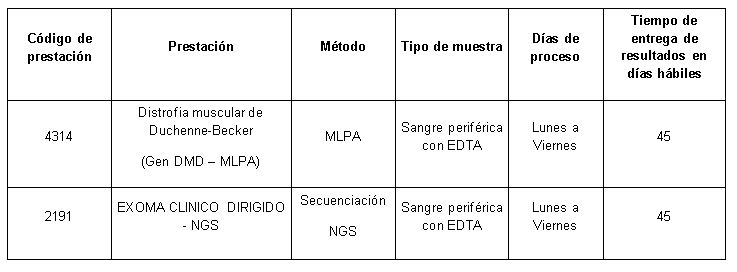
Determinaciones disponibles en Cibic:
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones.
Referencias
1 – Darras BT, Urion DK, Ghosh PS. Dystrophinopathies. 2000 Sep 5 [Updated 2018 Apr 26]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
2- Dooley J y cols. Duchenne muscular dystrophy: a 30-year population-based incidence study. Clin Pediatr (Phila). 2010. 49:177–9
3- Chelly J y cols. Effect of dystrophin gene deletions on mRNA levels and processing in Duchenne and Becker muscular dystrophies. Cell. 1990; 63:1239–48.
4- Abbs S. et al. (2010). Best practice guidelines on molecular diagnostics in Duchenne/Becker muscular dystrophies. Neuromuscul Disord. 20:422-7
Para mayor información o consultas:
 Sección: Biología Molecular
Sección: Biología Molecular
Lic. Alan Gomez.
Tel 0341-4722424. Interno: 243/225
 Sección: Biología Molecular
Sección: Biología Molecular
Lic. Analía Seravalle.
Tel 0341-4722424. Interno: 242.