

La Hemoglobinuria Paroxística Nocturna (HPN) o Síndrome de Machiafava-Michelli, es una patología crónica adquirida, poco frecuente (incidencia de 0,05-0,13 casos/ 100000 habitantes/ año), que se da como consecuencia de una expresión clonal, no maligna de células progenitoras hematopoyéticas que han adquirido una mutación somática en el gen PIG-A (brazo corto del cromosoma X). Este gen codifica para una enzima que cataliza el primer paso en la biosíntesis del grupo de anclaje glicosil-fosfatidil-inositol (GPI), condicionando así la carencia total o parcial de la expresión de proteínas ancladas a la membrana por GPI, tales como CD55 y CD59.
Esto lleva a la acción del complemento sobre los eritrocitos carentes de estas proteínas y la consecuente hemólisis. La intensidad de la hemólisis depende del tamaño del clon, el grado de alteración de las células y la activación del complemento.
A pesar de que esta mutación se manifiesta clínicamente sobre los eritrocitos, la misma también produce ausencia o disminución de la expresión de otras proteínas de membrana sobre los leucocitos: CD14 sobre monocitos y CD16, CD24 y CD66b sobre granulocitos.
En esta patología pueden distinguirse dos tipos de células: células HPN tipo II y HPN tipo III. Las células HPN tipo II son parcialmente deficientes en GPI (y sus proteínas asociadas) mientras que las células HPN tipo III presentan una deficiencia total.
A su vez, existen tres tipos de HPN:
– la HPN clásica, que presenta un clon en granulocitos cercano al 50 % y evidencias clínicas de hemólisis intravascular o trombosis sin evidencias de fracaso medular.
– la HPN en el contexto de otra patología hematológica, que presenta un clon en granulocitos, menor al 30 % y datos claros de hemólisis con otra patología medular asociada (anemia aplásica, SMD o mielofibrosis primaria, etc.)
– la HPN subclínica, la cual presenta un clon menor al 1% aunque sin datos de hemólisis y en general se da asociada a una aplasia medular.
El clon originado por esta célula mutada y toda su descendencia conserva su capacidad normal de proliferar y diferenciarse y no parece tener una inestabilidad genética ni capacidad de transformación diferente a la de sus contrapartes normales.
Las manifestaciones en esta enfermedad son la hemólisis intravascular caracterizada por anemia hemolítica crónica con episodios agudos (hemólisis nocturnas con hemoglobinurias matinales), la coexistencia muy frecuente de cierto grado de fallo medular y las trombosis, particularmente en sitios inusuales.
Una de las causas más importantes del bajo número de casos diagnosticados es la falta de conocimiento sobre la enfermedad que trae como resultado un diagnóstico tardío de la enfermedad, con el potencial desarrollo de complicaciones
Utilidad clínica de su análisis por Citometría de Flujo:
La Citometría de Flujo es el método de elección. Se utiliza para la detección y cuantificación de células con deficiencia de GPI, como también para la subclasificación del tipo celular.
Para el screening inicial de esta enfermedad, así como para evaluar el tamaño del clon, es preferible comenzar el análisis sobre las poblaciones de granulocitos y monocitos (pérdida parcial o total de CD16 y CD14, respectivamente) antes que sobre la de hematíes (pérdida parcial o total de CD59) ya que el análisis sobre estos últimos puede llegar a resultar confuso a causa de episodios de hemólisis y/o transfusiones previo al estudio.
Tipo de muestra, conservación y envío al laboratorio:
En todos los casos la muestra de elección para el análisis por citometría de flujo es sangre periférica anticoagulada con EDTA (puede usarse heparina o citrato), descartándose muestras de médula ósea, en las cuales la presencia de elementos inmaduros puede llevar a un diagnóstico erróneo.
La misma debe conservarse a temperatura ambiente y debe ser remitida al laboratorio dentro de las 24 h de extraída. En caso de no poder procesarse en ese lapso de tiempo, se debe agregar el estabilizante TransfixTM que conserva la muestra estable por 7 días.
Prestación disponible en Cibic Laboratorios
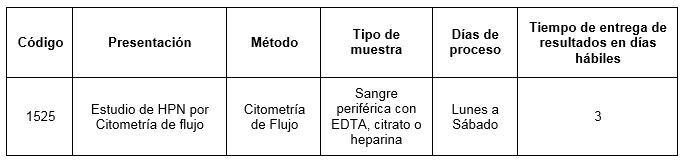
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Referencias:
1. Guidelines for the Diagnosis and Monitoring of Paroxysmal Nocturnal Hemoglobinuria and Related Disorders by Flow Cytometry- Borowitz et al. Cytometry Part B (Clinical Cytometry) 78B:211–230 (2010).
2. Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 2005; 106: 3699-709.
3. Hernández-Campo PM, Almeida J, Matarranz S, de Santiago M, Sánchez ML, Orfao A. Quantitative analysis of the expression of Glycosylphosphatidylinositol- anchored proteins during the maturation of different hematopoietic cell compartments of normal bone marrow. Cytometry 2007; 72B: 34-42.
4. Bioq. Nora Silvia Halperin. Diagnóstico de HPN (Rol de la Citometría de Flujo). Curso ABA 2017.
Para mayor información o consultas:
Sector: Citometría de Flujo
Bioq. Mariel Ambrosi
Teléfono: 0341-4722424. Interno: 259