
Las enfermedades cardiovasculares (ECV) conforman una de las mayores causas a nivel mundial de enfermedad y mortalidad, afectando a más de 16 millones de individuos anualmente. Tradicionalmente, el foco de atención ha sido puesto en lo que suelen denominarse factores de riesgo individuales asociados al estilo de vida (ej. dieta, tabaquismo, ejercicio) dado que las ECV se hallan frecuentemente relacionada a su vez, con otras enfermedades tales como diabetes y obesidad. Sin embargo, durante los últimos años, con el fin de diagnosticarlas en forma inequívoca, se ha prestado especial atención al estudio e identificación de factores de riesgo genéticos.
El Síndrome de QT Largo SQTL es una anormalidad estructural en los canales de potasio y sodio del corazón. Los síntomas incluyen arritmias ventriculares polimorfas, pudiendo desembocar en pérdidas de conciencia (síncopes), convulsiones, paros cardíacos y muerte súbita. Alrededor de 30% de los pacientes con SQTL pueden permanecer asintomáticos durante toda su vida, debiendo evitar una serie de fármacos que pueden alargar el QT (1).
El electrocardiograma de los pacientes se caracteriza por la prolongación del intervalo QT y además, con frecuencia, las ondas T tienen una morfología peculiar. El SQTL es una enfermedad hereditaria y tiene una prevalencia de aproximadamente una cada 5.000 personas, siendo una de las principales causas de muerte súbita en jóvenes. Actualmente, la presencia de mutaciones en tres genes han sido asociadas a aproximadamente el 75% de los casos: KCNQ1 (SQTL1), KCNH2 (SQTL2) y SCN5A (SQTL3).
Previamente, para el SQTL se diferenciaban sólo dos formas: I) SQTL asociada a sordera con herencia autosómica recesiva (Síndrome de Jervell-Lange-Nielsen, genes KCNQ1, KCNE1) ; II) SQTL no-asociado a sordera, con herencia autosómica dominante (Síndrome de Romano-Ward), este último asociado a mutaciones en los genes KCNQ1, KCNE1, KCNH2, KCNE2, SCN5A, CAV3, SCN4B, AKAP9, SNTA1, y KCNJ5 (2-4).
Es fundamental para este tipo de patologías el correcto diagnóstico molecular en el menor tiempo posible, impactando esto directamente en la toma de decisiones por parte de los médicos especialistas.
Las dificultades en el diagnóstico de mutaciones asociadas a CMH por técnicas de secuenciación tradicionales (tipo Sanger), radican en la elevada demanda en tiempo de mano de obra especializada debido al elevado número de genes que deben ser analizados. En la actualidad, es posible la realización de este tipo de estudios en forma paralela (ej. sobre la misma muestra, secuenciación simultánea de todos los genes sospechados) mediante secuenciación de segunda generación NGS (Next generation Sequencing, por sus siglas en Inglés).
Lo anteriormente mencionado impacta en forma directa en:
(i) el tiempo que demora la obtención de un correcto diagnóstico molecular siendo crítico el tiempo efectivo en el que un resultado es entregado al especialista solicitante.
(ii) costo-efectividad de el/los análisis a realizar (ej. la secuenciación de un único gen puede llevar varias semanas de trabajo, aumentando este tiempo si más de un gen debe incluirse en el pipeline del diagnóstico molecular).
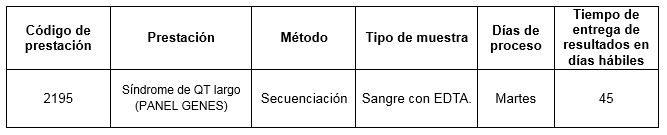
Prestación disponible en Cibic:
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Referencias
1- Ackerman MJ, y cols. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm Society (HRS); European Heart Rhythm Association (EHRA). Europace. 2011;13:1077-109.
2- Splawski I y cols. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–85.
3- Vatta M y cols. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114:2104–12.
4- Medeiros-Domingo A y cols. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation. 2007;116:134–42.
Para mayor información o consultas:
Sección: Biología Molecular

Dra. María Florencia Gosso.Teléfono: 0341-4499444. Int: 258.

Lic. Guadalupe Méjico. Tel: 0341-4499444. Int: 239.
 Lic. Analía Seravalle.Teléfono: 0341-4499444. Int: 242.
Lic. Analía Seravalle.Teléfono: 0341-4499444. Int: 242.