
La hemoglobina es una molécula compleja integrada por dos pares de cadenas polipeptídicas. Cada cadena está ligada a un hemo, un núcleo tetrapirrólico (porfirina) al que esta coordinado un átomo de hierro. La estructura hemo es común a todas las hemoglobinas y sus variantes. El tipo de hemoglobina está determinado por la fracción proteica, llamada globina. Las cadenas polipeptídicas α, β, δ y ɣ constituyen las hemoglobinas humanas normales.
En los adultos las siguientes variantes de hemoglobina son normales y componen el total de hemoglobina:
• Hemoglobina A (α2 β2): 95% a 98%
• Hemoglobina A2 (α2 δ2): 1,5 % a 3,5 %
• Hemoglobina F (α2 ɣ2): 0 % a 2%
La estructura espacial de la hemoglobina y otras de sus propiedades moleculares dependen de la naturaleza y la secuencia de los aminoácidos que constituyen las cadenas. La mayoría de hemoglobinopatías son debidas a la sustitución por mutación de un solo aminoácido en uno de los cuatro tipos de cadenas polipeptídicas. El significado clínico de tales cambios depende del tipo de aminoácido involucrado y de su posición. En enfermedades clínicamente significativas, pueden estar afectadas las cadenas α o β. Se han descrito más de 200 variantes de hemoglobina en adultos. Las primeras hemoglobinas anormales estudiadas y de aparición más frecuente tienen una carga eléctrica neta alterada, lo que facilita su detección por electroforesis. Existen cuatro hemoglobinas anormales principales que presentan un interés clínico particular: S, C, E y D.
En Cibic hemos incorporado recientemente el uso del kit HYDRAGEL HEMOGLOBIN de Sebia el cual, utilizado junto con el instrumento semiautomático HYDRASIS, permite la separación de las hemoglobinas normales (A y A2) y la detección de las principales variantes de hemoglobina: S o D y C o E mediante electroforesis en geles de agarosa tamponados alcalinos (pH 8,5). La lectura del gel mediante densitometría permite definir las concentraciones relativas (porcentajes) de cada fracción.
Patrones de migración posibles y su interpretación
Los kits Hydragel están destinados a la identificación preliminar de las hemoglobinopatías y talasemias. Una vez que se observe un patrón anormal, su identidad debería ser confirmada mediante pruebas discriminatorias apropiadas.
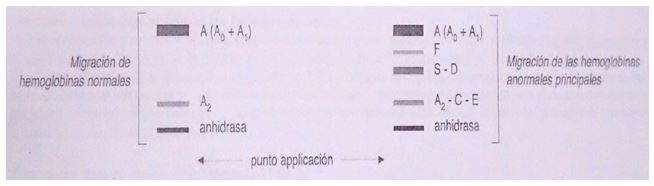
Figura: migración de bandas en gel de agarosa con tampón alcalino.
A0 y A1: fracciones no glicosiladas y glicosiladas de HbA normal de adulto.
Anormalidades cualitativas (hemoglobinopatías):
Hemoglobina S: es la más frecuente. Se produce por la sustitución de un ácido glutámico (aminoácido ácido) de la cadena β por una valina (aminoácido neutro). Esto da lugar a una enfermedad que puede ser muy grave, la anemia drepanocítica o anemia de células falciformes, nombre debido a que los eritrocitos tienen forma de hoz o media luna (drepanocitos). La anemia falciforme es frecuente en forma heterocigótica ya que en homocigosis es letal. Su movilidad electroforética se ve, por tanto, disminuida. En los geles de agarosa tamponados alcalinamente, la hemoglobina S migra entre las fracciones A y A2.
Hemoglobina C: un ácido glutámico de la cadena β está sustituido por una lisina (aminoácido básico) lo cual determina que su movilidad electroforética se vea fuertemente reducida. Esta mutación reduce la plasticidad normal de los eritrocitos causando una hemoglobinopatía. En aquellos que son heterocigotas para esta mutación, cerca del 28 a 44% de su hemoglobina (Hb) total es Hb C, y no desarrollan anemia. En los homocigotas, casi toda la Hb está en la forma de Hb C, dando lugar a una anemia hemolítica leve. En los geles C, E y A2 se superponen. Cuando esta fracción es > 15 % debe sospecharse la presencia de hemoglobina C y E.
Hemoglobina E: La Hb E es el resultado de una mutación en el gen que codifica la síntesis de la cadena β- globina, La síntesis de esta hemoglobina anormal se encuentra parcialmente reducida, por este motivo se relaciona con los síndromes β-talasémicos. Acostumbra a ser una enfermedad benigna. En tampón alcalino migra exactamente igual que la hemoglobina C.
Hemoglobina D: un ácido glutámico de la cadena β está sustituido por glutamina (aminoácido neutro). Esta hemoglobina migra en tampón alcalino exactamente igual que la hemoglobina S.
Anormalidades cuantitativas:
Talasemias: constituyen un tipo bastante heterogéneo de enfermedades genéticas caracterizadas por disminución en la síntesis de un tipo de cadena polipeptídica.
Hay dos tipos de síndromes talasémicos:
Talasemias alfa: se caracterizan por una disminución en la síntesis de las cadenas α.
El exceso de síntesis de las cadenas β y ɣ en relación a α induce la formación de tetrámeros sin cadena α:
Hemoglobina Bart: ɣ 4.
Hemoglobina H: β 4.
Talasemias beta: se caracterizan por descenso en la síntesis de cadenas β. Sólo la síntesis de Hemoglobina A se ve afectada, por lo tanto los porcentajes de Hb F y Hb A2 se incrementan con respecto a la Hb A.
Prestaciones disponibles en Cibic:

Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre la práctica consulte a la extranet o al manual de prestaciones.
Referencias
– V.F. Fairbanks, ed. (1980) Hemoglobinopathies and thalassemia: Laboratory methods and case studies. Brian C. Decker, New York.
– T.H.J. Huisman and J.H.P. Jonxis (1977) The hemoglobinopathies: techniques of identification. Marcel Dekker, New York.
– J.S. Krauss, P.A. Drew, M.H. Jonah, M. Trinh, S. Shell, L. Black and C.R. Baisden (1986) Densitometry and microchromatography compared for determination of the hemoglobin C and A2 proportions in hemoglobin C and hemoglobin SC disease and in Hemoglobin C trait. Clin. Chem. 32, 5, 860-863.
– R.G. Schneider (1978) Methods for detection of hemoglobin variants and hemoglobinepathies in the routine clinical laboratory. CRC Crit. Rev. Clin. Lab. Sci. 9, 243-271.
Para mayor información o consultas:

Sección: Producción Analítica No Automatizada
Bioq. Franco Delleppiagge
Teléfono: 0341 – 4499444. Interno: 220