
La osteogénesis imperfecta (OI) o “enfermedad de los huesos de cristal”, es un trastorno hereditario del tejido conectivo que afecta a la producción del colágeno, principalmente del tipo 1 y comprende un amplio espectro de presentaciones fenotípicas caracterizado por: baja masa ósea, fragilidad ósea, y amplio espectro en cuanto a su gravedad clínica. La variabilidad fenotípica va desde sujetos con huesos casi rectos y muy pocas fracturas a otros con múltiples fracturas. Por su baja incidencia, de 1/15.000 a 1/ 20.000 en recién nacidos, la OI pertenece al grupo de enfermedades raras, y afecta por igual ambos sexos, razas y grupos étnicos.
La mayoría de los casos de OI (90%) se originan por mutaciones heterocigotas (descriptas más de 1500) autosómico dominantes (AD) o de novo, en uno de los dos genes que codifican las cadenas pépticas de pro-colágeno I (COL1A1 y COL1A2). El colágeno tipo-1 es un componente estructural de la matriz extracelular del tejido conectivo, cuya función es proporcionar soporte y resistencia a la tracción a los tejidos. Esta proteína, la más abundante en hueso y piel, es sintetizada en el retículo endoplasmático en forma de molécula precursora tras el ensamblaje de dos cadenas peptídicas de pro-colágeno α1 (codificada por COL1A1) y otra de pro-colágeno α2 (codificada por COL1A2), en una triple hélice. La glicina se sitúa cada 3 residuos helicoidales (Gly-X-Y secuencia).En este proceso intervienen chaperonas moleculares y enzimas del retículo endoplasmático, las cuáles proporcionan las modificaciones postraduccionales (la hidroxilación de residuos específicos de prolina y lisina y la glicosilación de determinadas hidroxilisinas) necesarias para el correcto plegamiento de los trímeros de colágeno y su posterior “crosslinking” en la matriz extracelular.
Las anomalías genéticas más frecuentes encontradas en la OI-AD son mutaciones puntuales que afectan al residuo de glicina produciendo alteraciones en la estructura o en la cantidad de colágeno tipo 1, con un fenotipo esquelético y clínico que va desde subclínico a letal, dependiendo de la cadena que se vea afectada, en qué posición de la triple hélice se produce la sustitución y del aminoácido que sustituye a la glicina.
Los restantes casos de OI (10%) son autosómico recesivos (AR) y se caracterizan por una elevada heterogeneidad genética. Entre los genes de OI-AR descriptos hasta la fecha se encuentran las tres enzimas que forman el complejo de hidroxilación de la Prolina 986 de la cadena de procolágeno a1 (CRTAP, LEPRE1 y PPIB); las chaperonas FKBP65 (codificada por FKBP10) y HSP47 (codificada por SERPINH1), que intervienen en el plegamiento y secreción del procolágeno I; SERPINF1, un factor secretable que interacciona con la matriz extracelular y con función anti-angiogénica; y TMEM38B, un canal específico de cationes monovalentes involucrado en liberar Ca(2+) de los reservorios intracelulares.
El área de Biología Molecular de Cibic ha incorporado recientemente las siguientes prestaciones:
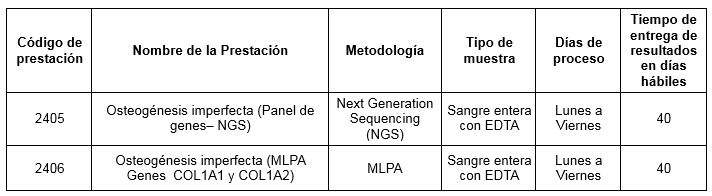
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte a la extranet o al manual de prestaciones.
Bibliografía
– Rauch F & Glorieux FH. Osteogenesis Imperfecta. Lancet 2004; 363:1377-1385.
Para mayor información o consultas:
Sección: Biología Molecular
Dra. María Sonia Baquedano. Tel: 0341-4499444. Int: 242
Bioq. María Florencia Gosso, PhD. Tel: 0341-4499444. Int: 258