

El ácido oxálico es un ácido dicarboxílico que proviene principalmente del metabolismo endógeno y solo una pequeña parte de la dieta. Se produce en el hígado a partir del glioxalato y contribuye al 60-80% del oxalato plasmático. La mayor parte del oxalato ingerido se une en el tracto intestinal al calcio, si está disponible, o es degradado por bacterias que degradan el oxalato. Sólo se absorben en el tracto intestinal pequeñas cantidades de oxalato dietético. Su principal ruta de excreción es el riñón.
La hiperoxaluria ocurre debido a una incrementada concentración sérica y una incrementada carga filtrada renal de oxalato debido a diferentes causas. Las mismas se detallan en la Tabla 1.
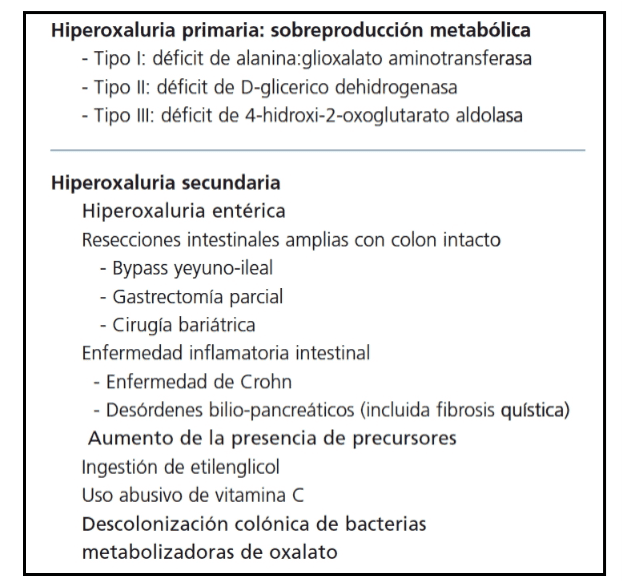
Tabla 1. Clasificación de los estados hiperoxalúricos.
La hiperoxaluria primaria es un desorden genético de herencia autosómica recesiva con incrementada producción endógena de oxalato.
El trastorno más frecuente y grave se debe al déficit enzimático de alanin glioxalato aminotransferasa (HOP tipo I) específico en el peroxisoma hepático (ver Figura 1). Dado que el oxalato no se metaboliza en los humanos y se elimina por vía renal, el riñón es el primer órgano afectado, dando lugar a la aparición de litiasis a repetición, nefrocalcinosis e insuficiencia renal precoz. Con la progresión de la insuficiencia renal, el oxalato cálcico se deposita masivamente en los tejidos (oxalosis).
La Hiperoxaluria tipo 2 (HOP-II) se debe a un defecto en el gen GRHPR que resulta en la deficiencia de la enzima hidroxipiruvato reductasa; y la Hiperoxaluria tipo 3 (HOP-III) es debida a mutaciones recesivas en el gen HOGA1.
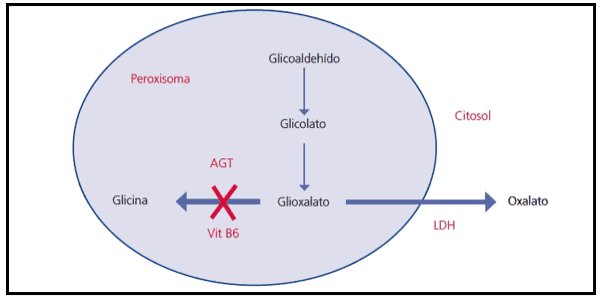
Figura 1: Metabolismo del ácido oxálico. AGT: alanin glioxalato aminotransferasa.
Ciertas patologías intestinales inflamatorias, como la Enfermedad de Crohn y otras derivadas de resecciones intestinales, pueden derivar en hiperoxaluria entérica. Normalmente, el calcio ingerido forma complejos con el oxalato y, en consecuencia, no está disponible para la absorción. En estos pacientes, el calcio se une a los ácidos grasos mal absorbidos en lugar de al oxalato, por lo que queda más oxalato libre y disponible para la absorción.
La cirugía de bypass gástrico también puede derivar en problemas de absorción de grasas en el intestino, lo que aumenta el riesgo de hiperoxaluria.
Otros factores que conducen a una mayor absorción de oxalato en el tracto intestinal es la deficiencia o ausencia total de bacterias intestinales que degradan el oxalato (principalmente Oxalobacter formigenes y Pseudomona oxalacticus); y la ingesta de dosis altas de vitamina C, ya que el ácido ascórbico se metaboliza a oxalato.
El patrón de presentación de la patología es muy heterogéneo. Las manifestaciones clínicas destacadas son nefrolitiasis recidivante, nefrocalcinosis, infecciones urinarias, hematuria e insuficiencia renal de rápida evolución. Las formas más graves aparecen en la HOP-I, habiéndose descrito formas infantiles que debutan en los primeros meses de vida. Las más comunes aparecen en torno a la segunda década de la vida y muchos pacientes se tratan como una litiasis recidivante. Existen variantes menos agresivas que se diagnostican en la edad adulta por la presencia de litiasis y/o nefrocalcinosis. La oxalosis o hiperoxaluria primaria en su etapa terminal puede provocar diversas complicaciones extra-renales: enfermedades de los huesos, anemia, problemas cardíacos y de la visión, alteración en el crecimiento de los niños, entre otros.
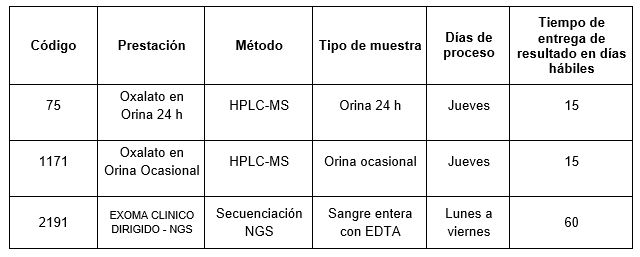
En CIBIC Laboratorios realizamos la determinación de Oxalato en Orina de 24 hs y Oxalato en Orina ocasional por cromatografía líquida de alta resolución a través del cromatógrafo Waters Alliance 2695 acoplado a un detector Qda (MASA SIMPLE).
A su vez, en caso de existir una fuerte sospecha de una etiología genética, es importante indagar aquellos genes implicados en el metabolismo del oxalato para la búsqueda de variantes patogénicas bialélicas que puedan explicar el cuadro. Esta determinación se puede realizar mediante un estudio de Exoma clínico dirigido-NGS aplicando la técnica de secuenciación de segunda generación (NGS).
Prestaciones disponibles en Cibic Laboratorios:
Es fundamental que la orina se recoja en medio ácido o sea acidificada en las primeras 24 horas, llegando a un pH <2.
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Referencias
1- Lorenzo, V; Torres, A; Salido, E. Primary hyperoxaluria. Revista Nefrología (Madr.) vol.34 no.3.Cantabria. 2014
2- Bele, U; Hajdinjak, T. El papel del oxalato en la urolitiasis. WebMed Central. 2012
3- Orozco, R; Camaggi, M. Evaluación metabólica y nutricional en litiasis renal. Revista médica clínica Las Condes. Vol XXI. Num 4. 2010
4- Lorenzo, V; Torres A. Diagnóstico y tratamiento de la hiperoxaluria primaria. NEFROLOGIA. Vol. XVI. Núm. 2. 1996
Para mayor información o consultas:
Sección: Química Analítica
Bioq. Corina Santos
Tel: +54 (341) 4861600. Int: 262