
Es una enfermedad asociada a una microdeleción intersticial que como pasa con otros síndromes de microdeleciones, puede también seguir un patrón de herencia autosómico dominante (1).
La incidencia es de 1 en 7500–20.000 nacidos vivos y afecta ambos sexos por igual. Se origina por deleción de una región crítica (~1.5 Mb) en el brazo largo del cromosoma 7 en el locus q11.23 (1, 2).
Los genes involucrados en la deleción de la región crítica incluyen a CLIP2, ELN, GTF2I, GTF2IRD1 y LIMK1. El gen ELN codifica para la proteína elastina. Esta proporciona la fortaleza y elasticidad a las paredes de los vasos sanguíneos, por lo que la pérdida del gen ELN está asociado con anormalidades del tejido conjuntivo y trastornos cardiovasculares. La deleción en los genes CLIP2, GTF2I, GTF2IRD1, LIMK1 y quizás otros genes estaría asociada a dificultades espacio-visuales, un comportamiento característico y otras dificultades cognitivas (2, 3).
Los niños con este síndrome, entre otras características clínicas, presentan facies característica de “duendecillo” o “elfica”. Es frecuente una excesiva sensibilidad a los estímulos auditivos o hiperacusia. Se observa retraso del crecimiento pre, posnatal y psíquico de variable intensidad. Es común la presencia de malformaciones cardíacas como estenosis aórtica supravalvular y valvular, coartación de la aorta, entre otras.
Una de las características típica de este síndrome es que los individuos afectados por este síndrome tienden a ser extremadamente amistosos y comunicativos. Los ojos presentan estrabismo interno, son azules con el iris estrellado, a veces, con opacidades corneales o del cristalino. Algunos niños de temprana edad tienen altos los niveles de calcio en la sangre. La hipercalcemia puede causar una irritabilidad extrema y síntomas similares a los cólicos. Además de alteración de los niveles de paratohormona y calcitonina debido a la hipercalcemia (1, 2, 3).
La deleción en la mayoría de los casos no puede ser observada por bandeo G, ni aún por cariotipo de alta resolución con niveles de relación de 850 bandas, pero puede ser detectada habitualmente por técnicas moleculares de FISH (figura 1) o microarray. Estas se emplean para confirmar o llegar al diagnóstico de este tipo de enfermedades (3).
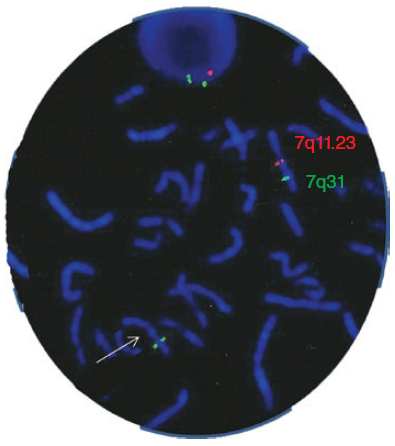
Figura 1. Análisis de FISH en metafase de un caso con Síndrome Williams: ish del(7)(q11.23q11.23)(ELN-). Adapataddo de Cytogenetic abnormalities : chromosomal, FISH, and microarray-based clinical reporting. Susan Mahler Zneimer. (2014). Wiley Blackwell.
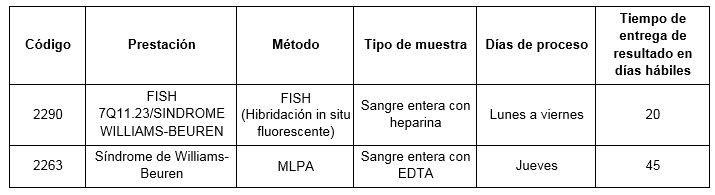
Prestaciones disponibles en Cibic:
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Referencias:
1. Arsham MS, Barch, MJ Lawce HJ. (2017). The AGT Cytogenetics Laboratory Manual. 4th Edition. Wiley-Blackwell.
2. Gersen SL, Keagle MB. (2013). The Principles of Clinical Cytogenetics. USA. Humana Press.
3. Zneimer. S. M. (2014) Cytogenetic Abnormalities: Chromosomal, FISH, and Microarray-Based Clinical Reporting and Interpretation of Result. Wiley-Blackwell, UK
4. Hibridación in situ Fluorescente (FISH). https://www.cibic.com.ar/laboratorios-bioquimicos/hibridacion-in-situ-fluorescente-fish/
Para mayor información o consultas:
Sección: Citogenética.
Dr. Carlos Zumoffen.
Tel: 0341-4499444. Interno 283.
Sección Biología Molecular
Dra. María Florencia Gosso. Tel: 0341 4499444. Int: 258
Dra. Maria Fernanda Madeira. Tel: 0341 4499444. Int: 239
Dra. Ivana Canonero. Tel: 0341 4499444 Int: 258/239
Lic. Guadalupe Méjico. Tel: 0341 4499444. Int: 239
Lic. Analía Seravalle. Tel: 0341 4499444. Int: 242