

El Síndrome de Noonan (SN, OMIM#163950) (1) es un trastorno genético relativamente común, su prevalencia es 1/1000-2500 nacidos vivos. Se hereda de forma autosómica dominante, donde un significativo porcentaje de pacientes presenta mutaciones de novo y en un 30-75% de individuos se reconoce un padre afectado.
El diagnóstico clínico se basa en una apariencia facial característica (hipertelorismo ocular, fisuras palpebrales descendentes, ptosis, orejas bajas y rotadas posteriormente con helix grueso), retraso de crecimiento posnatal, baja talla, defectos cardíacos congénitos (estenosis de la válvula pulmonar, defecto septal atrial y cardiomiopatía hipertrófica), retraso mental variable en un 25% de los casos, cuello corto y ancho, defectos esqueléticos (anomalías de esternón y columna, cúbito valgo), criptorquidia y trastornos de la coagulación (2).
SN presenta heterogeneidad genética. En el 30-60 % de los casos se encuentra asociado a mutaciones de tipo missense en el gen PTPN11 (OMIM*176876) localizado en el brazo largo del cromosoma 12 (12q24) (3). La mayoría de las mutaciones son recurrentes y están localizadas en los exones 2, 3, 8, 9 y 13 del gen PTPN11 (4).
Existen otros genes con mutaciones reportadas que han sido asociadas al Síndrome de Noonan: en aproximadamente un 13% de los casos se han hallado mutaciones en el gen SOS1 (OMIM*182530); en un 5% de los casos se han identificado mutaciones en el gen RAF1 (OMIM*164760); en menos del 5% mutaciones en los genes KRAS (OMIM*190070) y en menos de 2% BRAF (OMIM*164757) y NRAS (OMIM*164790).
Mutaciones en otros genes (CBL, MAP2K1, MAP2K2, SHOC2, HRAS) también han sido asociadas a Síndrome de Noonan-like o Rasopatías.
Rasopatías es el término empleado para el grupo de síndromes con anomalías del desarrollo que resultan de un mecanismo patogénico común: la desregulación de la vía Ras/MAPK, entre ellos SN. Todos estos genes están implicados la regulación de la vía Ras/MAPK.
Dada la alta prevalencia de mutaciones en los exones 2, 3, 8, 9 y 13 del gen PTPN11, ante un diagnóstico clínico presuntivo para Síndrome de Noonan, se recomienda el screening de mutaciones en el gen PTPN11 como primera línea. Si este estudio, fuera negativo y la sospecha clínica continúa, se debería continuar en segunda línea con la secuenciación del panel de genes asociados a Síndrome de Noonan-like, el cual podrá ayudar a establecer el diagnóstico diferencial entre las diferentes rasopatías o familia de síndromes neuro-cardio-facio-cutáneos.
Prestaciones disponibles en Cibic:
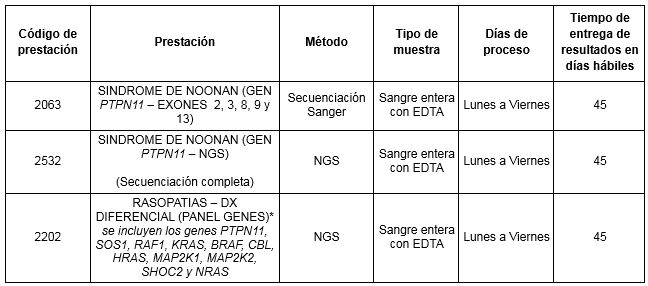
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte a la extranet o al manual de prestaciones.
Referencias
1. http://www.omim.org/
2. Tartaglia M. Y cols. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am. J. Hum. Genet. 70: 1555-1563, 2002.
3. Van der Burgt I y cols. Clinical and molecular studies in a large Dutch family with Noonan syndrome. Am. J. Med. Genet. 53: 187-191, 1994.
4. Tartaglia M y cols. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nature Genet. 29: 465-468, 2001.
Para mayor información o consultas:
Sección Biología Molecular
Lic. Guadalupe Méjico – gmejico@cibic.com.ar
Bioq. Ma. Florencia Gosso, PhD – mfgosso@cibic.com.ar
Dra. Nadia Cambados – ncambados@cibic.com.ar
Tel: 0341 – 4499444 internos 239 / 258.