

La leucemia mieloide crónica (LMC) es un síndrome mieloproliferativo crónico de naturaleza clonal, originada en las células madre hematopoyéticas, que resulta en un excesivo número de células mieloides en todos los estadios de maduración. Fue la primera enfermedad maligna en que se demostró una anomalía genética adquirida y es, en la actualidad, el modelo molecular de leucemia mejor estudiado (1).
Se caracteriza por la presencia del cromosoma Philadelphia (Ph), que resulta de la translocación recíproca entre los cromosomas 9 y 22 [t(9;22)(q34;q11)], y genera la yuxtaposición de los genes BCR y ABL1 dando origen a una proteína oncogénica con actividad de tirosina quinasa incrementada, alterando las vías de proliferación y supervivencia.
Según el punto de ruptura de los genes BCR y ABL1, se generan distintos rearreglos (b2a2 o b3a2, e1a2 y e19a2), dando lugar a proteínas de distintos pesos moleculares (P210, P190, P230). En la mayoría de las LMC, se puede detectar el transcripto de la isoforma P210, pero se han descripto casos con P190, P230 u otras con menor frecuencia.
El mejor conocimiento de la biología de la enfermedad y la descripción de los mecanismos de resistencia, permitió el desarrollo de tratamientos blanco-moleculares como inhibidores de tirosina quinasa (ITK), logrando una ventaja significativa en la sobrevida de estos pacientes, dada la gran efectividad en la inactivación de la proteína oncogénica. De esta manera, la introducción del imatinib, generó un cambio en el seguimiento de la LMC.
La enfermedad se identifica mediante un hemograma y frotis de sangre periférica que revela hiperleucocitosis con marcada desviación a la izquierda. Puede haber esplenomegalia.
La confirmación del diagnóstico se obtiene por la identificación del cromosoma Ph (estudio citogenético por bandeo G), FISH o PCR para BCR-ABL1. La RT-PCR informa el tipo de transcripto presente en cada paciente (b2a2 o b3a2 u otras) y la qRT-PCR (opcional en el momento del diagnóstico) permite conocer el nivel basal de transcriptos y es el método recomendado para el seguimiento de la enfermedad residual.
El estudio citogenético por bandeo G se realiza en médula ósea y es una metodología que tiene alta especificidad y baja sensibilidad. Se recomienda su realización al momento del diagnóstico y hasta que alcance la remisión citogenética completa. Para este estudio, es importante disponer de una buena calidad de médula ósea (1-2 ml del primer aspirado) colectada con heparina y obtenida en forma estéril (dado que cualquier contaminación impide el desarrollo celular).
En el 95% de los casos se detecta la t(9;22)(q34;q11), denominada translocación clásica. En el 5% restante el cromosoma Ph puede no detectarse aun ocurriendo la fusión BCR-ABL (Ph enmascarado) o resultar de translocaciones variantes, involucrando a otro/s cromosomas además de los cromosomas 9 y 22 (variantes crípticas de la translocación). En este caso, la confirmación del diagnóstico depende de la detección del gen de fusión BCR-ABL1 por otros métodos más específicos y sensibles como FISH y PCR. Estos pacientes se tratan de la misma manera que los pacientes Philadelphia positivos (Ph+).
Al momento del diagnóstico y durante el seguimiento, el estudio citogenético se realiza evaluando entre 10-20 metafases, para poder definir correctamente el tipo de respuesta citogenética alcanzada.
Cuando el número de metafases es insuficiente (o no se pudo detectar Ph) los mismos extendidos citogenéticos pueden ser procesados para la técnica de FISH. En la LMC se ha observado que el clon Ph+ puede adquirir nuevas alteraciones citogenéticas además del clásico cromosoma Ph lo cual se denomina evolución clonal. Este mecanismo se asocia con evolución de la enfermedad (2).
El FISH o Hibridación In Situ Fluorescente es una técnica que detecta secuencias de ADN en células o tejidos preservados mediante el empleo de una sonda (fragmento de ADN homólogo a la secuencia blanco) marcada con un fluorocromo, la cual va dirigida hacia un lugar específico del ADN y que emite fluorescencia que puede ser observada por medio de un microscopio de epifluorescencia. La misma se fundamenta en la capacidad que poseen los ácidos nucleicos para hibridarse entre sí, es decir, la existencia de determinada secuencia de ADN, que resulta complementaria con otra secuencia (3).
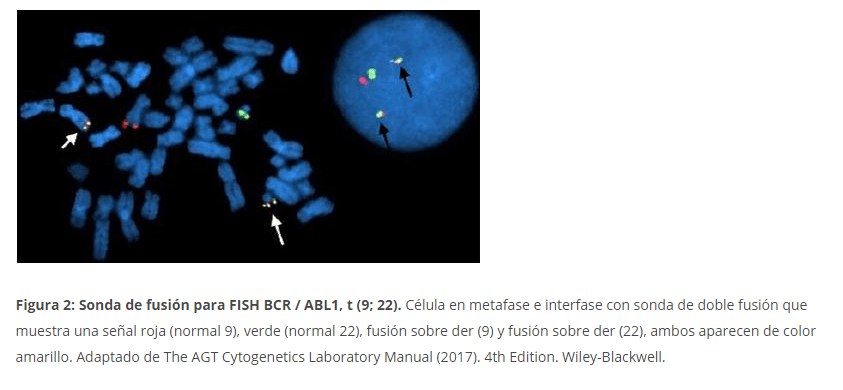
Para detectar la translocación BCR- ABL se utiliza una sonda de tipo Doble color/Doble fusión, la cual detecta la unión de dos genes normalmente separados, ya sea por translocación o deleción intersticial de fragmentos cromosómicos.
Dos genes involucrados en translocaciones frecuentes son hibridados con sondas de diferente color. Además del gen target, las sondas abarcan varios Kb en dirección 3’ y 5’. De esta forma al ocurrir la translocación, parte de la sonda queda en el cromosoma original y parte va en el derivado. Al combinarse con la sonda proveniente del otro cromosoma involucrado, se generan 2 señales de fusión.
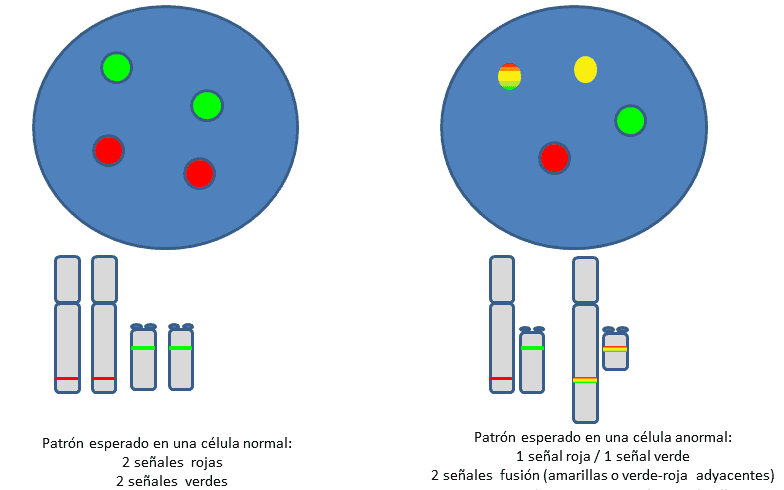
Su principal ventaja es que puede aplicarse sobre células en interfase y facilita su análisis sobre un mayor número de células, además de ser una técnica más sensible y especifica (4).
Prestaciones disponibles en Cibic Laboratorios:
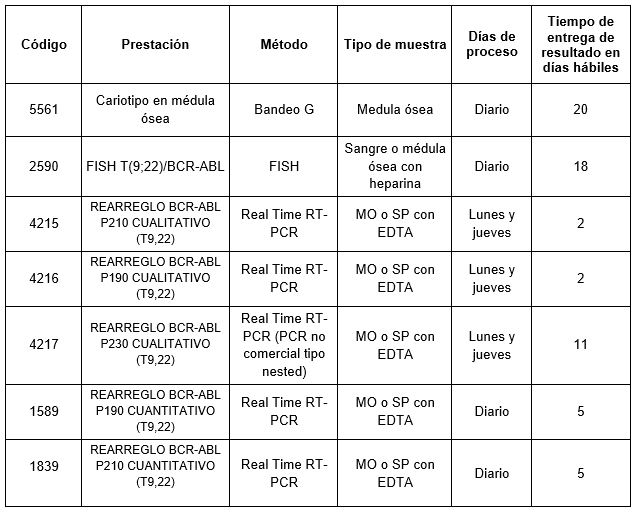
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Referencias
1. Pavón Morán Valia y cols. Leucemia mieloide crónica. Actualización en citogenética y biología molecular. Vol. 21 Núm.2. Mayo – Agosto 2005, Rev Cubana Hematol Inmunol Hemoter 2005.
2. Beligoy, Luis Bendek y cols. Guía de Diagnóstico y Tratamiento. Sociedad Argentina de Hematología. Pág. 461, 2019.
3. Gersen SL, Keagle MB. The Principles of Clinical Cytogenetics. USA. Humana Press. 2013.
4. Daniel A. Arber y cols. La revisión de 2016 de la clasificación de la Organización Mundial de la Salud de neoplasias mieloides y leucemia aguda. 2016
Para mayor información o consultas:
Sección: Citogenética
 Bioq. Sibila Bertalot
Bioq. Sibila Bertalot
 Lic. Veronica Vanrell
Lic. Veronica Vanrell
 Tec. Victorina Carbone
Tec. Victorina Carbone
Tel: 0341-4861600. Int: 283