

Disgenesia gonadal (DG) es un término utilizado para un subconjunto de trastornos del desarrollo sexual (DSD, por sus siglas en ingles). Se caracteriza por una formación incompleta o defectuosa de las gónadas (ovario o testículo) debido a anomalías estructurales o numéricas de los cromosomas sexuales o mutaciones en los genes implicados en el desarrollo de las gónadas (1,2).
Las gónadas disgenéticas están asociadas a un riesgo alto (30-60 %) de desarrollar tumores abdominales. Los pacientes con DG que tienen un cromosoma Y o material del cromosoma Y, tienen un mayor riesgo de desarrollar tumores de células germinales como gonadoblastoma o carcinoma in situ (CIS), con el potencial de transformación maligna a disgerminoma o seminoma (3).
Dentro de las disgenesias gonadales se incluye el síndrome de Turner y sus variantes, la disgenesia gonadal pura (DGP) 46XX o disgenesia gonadal mixta (DGM) 46XY, y el hermafroditismo verdadero (4). Por su parte las DGP se clasifican en 46,XX o 46,XY según el sexo cromosómico, y en parciales o completas según el grado de diferenciación gonadal. Dentro de las DGP con cariotipo 46,XY; la falta completa de diferenciación testicular constituye la disgenesia gonadal pura completa 46,XY; llamada también Síndrome de Swyer (4).
El Síndrome de Swyer (OMIM: 400044) es un trastorno del desarrollo sexual que se caracteriza por presentar un fenotipo femenino, a pesar de un cariotipo 46,XY. Presentan genitales externos femeninos normales, pero con falta de desarrollo puberal. Se ha observado que aproximadamente en el 15-20% de los pacientes, el síndrome de Swyer se produce debido a mutaciones del gen SRY (gen determinante del sexo en el cromosoma Y) o la eliminación del segmento del cromosoma Y que contiene el gen SRY.
El gen SRY está situado en el brazo corto del cromosoma Y (Yp11.3) y es un factor crítico para iniciar la determinación del sexo masculino al activar el tejido gonadal no diferenciado para que se transforme en testículos. La ausencia o mutación de este gen, que son en su mayoría de novo aunque algunos individuos heredan el gen mutado, hace que los testículos no se formen (3).
El diagnóstico del Síndrome de Swyer se establece en base al examen físico, la evaluación hormonal, estudios de imagen y estudios genéticos que incluyen el cariotipo, la histología gonadal y determinaciones por biología molecular (3).
Los métodos moleculares, que permiten detectar presencia o ausencia del gen SRY, así como la secuenciación del mismo que permite evidenciar mutaciones, constituyen herramientas útiles en el contexto de la evaluación clínica ante la sospecha de Síndrome de Swyer al presentar una alta sensibilidad y especificidad.
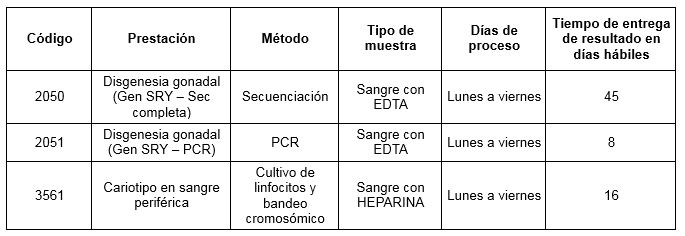
Prestaciones disponibles en Cibic:
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Referencias
1- Lee PA, Houk CP, Ahmed SF: Consensus statement on management of intersex disorders: international consensus conference on intersex. Pediatrics 2006, 118:e488–e500
2- MacLaughlin DT, Donahoe PK: Sex determination and differentiation. N Engl J Med 2004, 350:367–378
3- State of the art review in gonadal dysgenesis: challenges in diagnosis and management McCann-Crosby et al. International Journal of Pediatric Endocrinology 2014, 2014:4 Page 2 of 17.
4- C. Torreira Banzas, A. Repáraz Andrade. DISGENESIAS GONADALES Caso Clínico: Síndrome de Swyer. GENÉTICA MOLECULAR APLICADA AL DIAGNÓSTICO DE ENFERMEDADES HEREDITARIAS 2013-2014 Ed Cont Lab Clín; 18: 36 – 44.
Para mayor información o consultas:
 Sección: Biología Molecular
Sección: Biología Molecular
Lic. Alan Gomez
Tel: 0341-4499444. Int: 240