

Las enfermedades mendelianas (EM) conforman un grupo de patologías hereditarias heterogéneas con un amplio espectro fenotípico. Las EM se hallan asociadas, en su mayoría, a mutaciones puntuales del ADN localizadas en regiones codificantes de genes cuya función es esencial para la regulación y mantenimiento de procesos biológicos en el organismo.
Caracterizadas por una alta heterogeneidad genética, es fundamental para este tipo de patologías el correcto diagnóstico molecular en el menor tiempo posible, impactando esto directamente en la toma de decisiones por parte de los médicos especialistas y también en la disminución de costos de hospitalización sin un diagnóstico clínico asociado.
Las dificultades en el diagnóstico de mutaciones asociadas a EM por técnicas de secuenciación tradicionales (tipo Sanger), radican en la elevada demanda en tiempo de mano de obra especializada debido al elevado número de secuencias que deben ser analizadas en forma “secuencial” (valga la redundancia), sin tener la posibilidad de realizar este tipo de estudios en forma paralela (ej. sobre la misma muestra, secuenciación simultánea de todos los genes sospechados).
En muchas ocasiones la chance de hallar mutaciones causales para el diagnóstico de una determinada EM se halla asociada al estudio de varios genes, siendo frecuentemente, debido la naturaleza de la patología, prácticamente imposible definir el orden secuencial en que estos genes deberían ser investigados (ej. inicio del diagnóstico molecular por aquellos genes más frecuentemente asociados a la patología dejando para etapas posteriores genes menos frecuentemente asociados). Todo ello impacta en forma directa en: (i) el tiempo que demora la obtención de un correcto diagnóstico molecular siendo crítico el tiempo efectivo en el que un resultado es entregado al especialista solicitante, (ii) costo-efectividad de el/los análisis a realizar (ej. la secuenciación de un único gen puede llevar varias semanas de trabajo, aumentando este tiempo si más de un gen debe incluirse en el diagnóstico molecular).
Es por todo lo anteriormente mencionado que el análisis genético del exoma (exoma clínico) mediante el uso de la tecnología de secuenciación masiva NGS (también llamada de alto rendimiento o de última generación), la cual permite secuenciar en forma simultánea un número elevado de muestras de pacientes para múltiples genes, se ha convertido en una herramienta fundamental para el diagnóstico molecular de enfermedades Mendelianas, altamente heterogéneas, tanto a nivel pediátrico como neonatal (1,2).
Es posible realizarlo a través de “Exoma clínico dirigido”, en pacientes con sospecha clínica para una enfermedad o síndrome clínicamente definido. Sin embargo, fundamentalmente en pacientes con desórdenes del espectro autista, déficit intelectual no sindrómico y malformaciones congénitas múltiples se recomienda el estudio de secuenciación de “Exoma clínico en trío” (paciente o probando, padre y madre) que permite analizar la totalidad de las variantes presentes en el panel de Exoma clínico (más de 4.800 genes) sin dirigir el estudio a paneles de genes candidatos.
El análisis de Exoma clínico en trio tiene la ventaja, al incorporar la secuenciación de las muestras de los progenitores, de facilitar con el análisis concurrente la selección e interpretación, de manera de priorizar aquellas variantes codificantes, de novo o heredadas (autosómicas dominantes, recesivas y ligadas al cromosoma X) con potencial relevancia clínica. Otra utilidad es permitir revelar nuevos genes candidatos responsables de la patología con probable potencialidad terapéutica.
Se destaca que al igual que los estudios de exoma clínico dirigido la información clínica del paciente es fundamental para dar el soporte de que la variante identificada es la responsable del fenotipo del paciente (1,2).
Prestaciones disponibles en Cibic:
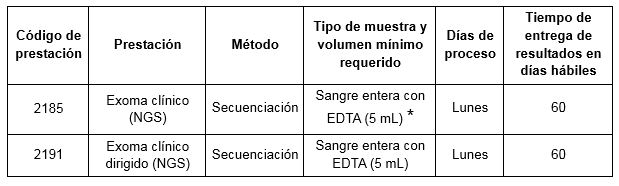
* Para la realización de este estudio se requiere muestras de progenitores
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones.
Referencias
1. Lee H y cols. Clinical Exome Sequencing for Genetic Identification of Rare Mendelian Disorders. JAMA. 2014; 312: 1880–1887.
2. Meng L y cols. Use of Exome Sequencing for Infants in Intensive Care Units: Ascertainment of Severe Single-Gene Disorders and Effect on Medical Management. JAMA Pediatr. 2017;171:e173438
Para mayor información o consultas:
Sección: Biología molecular
 Dra. María Fernanda Madeira Tel: 0341-4499444. Int: 239
Dra. María Fernanda Madeira Tel: 0341-4499444. Int: 239
 Dra. María Florencia Gosso. Tel: 0341-4499444. Int: 258
Dra. María Florencia Gosso. Tel: 0341-4499444. Int: 258
 Lic. Analía Seravalle. Tel: 0341-4499444. Int: 242
Lic. Analía Seravalle. Tel: 0341-4499444. Int: 242
 Lic. Guadalupe Méjico. Tel: 0341-4499444. Int: 239
Lic. Guadalupe Méjico. Tel: 0341-4499444. Int: 239