
El Síndrome MELAS del ingés “Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes”, y en español “Encefalomiopatía mitocondrial, acidosis láctica y episodios parecidos a un accidente cerebrovascular”, es un desorden multisistémico, con un fenotipo clínico variable que comienza típicamente en la niñez, entre los 2 y 10 años.
El desarrollo psicomotor temprano es habitualmente normal. La afectación del sistema nervioso central puede manifestarse inicialmente con convulsiones tónico-clónicas, cefaleas recurrentes, anorexia y vómitos. Episodios simil stroke, son accidentes cerebrovasculares que pueden ser recurrentes con pérdida de conciencia, hemiparesia y hemianopsia, afectan gradualmente las habilidades motoras, intelectuales y visuales.
Una manifestación inicial también puede ser la debilidad muscular proximal de miembros o intolerancia al ejercicio. La pérdida auditiva sensorioneural es un hallazgo común. La enfermedad es producida por presencia de variantes patogénicas en el ADN de las mitocondrias, organelas que se ubican en el citoplasma celular y es transmitida por vía materna a sus descendientes, siendo la madre sintomática o no.
El diagnóstico de síndrome de MELAS se basa en la combinación de hallazgos clínicos y en la determinación de variantes patogénicas en el ADN mitocondrial (mtADN). Mutaciones en el gen MT-TL1 son las más frecuentemente asociadas a la patología, la más común presente en el 80% de los casos es una transición de una Adenina por Guanina en el nucleótido 3243 (m.3243 A>G) y otras mucho menos frecuentes en el mismo gen son (m.3271 T>C) y (m.3252 A>G).
Debido a la ocurrencia de heteroplasmia, es decir a la presencia de ADN mitocondrial normal y mutado en la misma célula y a su diferente proporción en distintos tejidos del organismo, la enfermedad tendrá una expresión y severidad variables.
Esto también explicaría que en ocasiones las variantes patogénicas no sean halladas en leucocitos de sangre periférica, debiendo estudiarse otros tejidos, como cultivo de fibroblastos de piel, musculo esquelético y otros. En caso de no hallar alguna de éstas tres variantes patogénicas en el gen MT-TL1 asociadas a la enfermedad, deberá completarse con la secuenciación de genes conocidos de causar MELAS o realizar la secuenciación completa del genoma mitocondrial.
Prestación disponible en Cibic:
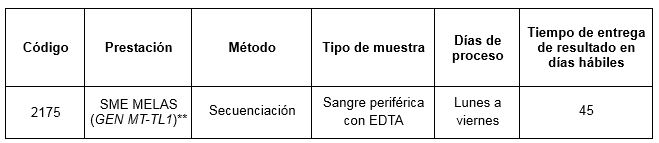
** Estudio de mutaciones m.3243A>G, m.3252A>G y m.3271T>C en el gen mitocondrial MT-TL1 por secuenciacion Sanger.
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Bibliografía:
1. OMIM (https://omim.org/)
2. Genereview (https://www.ncbi.nlm.nih.gov/books/NBK1233/)
Para mayor información o consultas:
Sector: Biología Molecular
 Lic. Analía Seravalle Tel: 0341 4499444 Int: 242
Lic. Analía Seravalle Tel: 0341 4499444 Int: 242
 Dra. María Florencia Gosso Tel: 0341 4499444 Int: 258
Dra. María Florencia Gosso Tel: 0341 4499444 Int: 258