

La Homocisteína es un aminoácido no esencial que deriva del aminoácido esencial, metionina, que proviene de la ingesta (dieta), principalmente de proteínas animales. La diferencia entre estos dos aminoácidos está, en que la metionina posee un grupo metilo (CH3) adicional, que la convierte en el principal “donante de grupos metilos”. La metionina llega al hígado a través del sistema portal, una vez allí, se utiliza para la síntesis de proteínas o se transforma en Homocisteína. En la “desmetilación hepática” de la metionina para el paso a Homocisteína interviene la metionina adenosiltransferasa (MAT), una enzima clave que regula la obtención de radicales metilos necesarios para el organismo.
Los trastornos en el metabolismo de la homocisteína ocasionan hiperhomocisteinemia (concentraciones elevadas de homocisteína en suero o plasma) y/u homocistinuria (las concentraciones elevadas en plasma provocan que la homocisteína tenga que excretarse por la orina). La hiperhomocisteinemia está causada por deficiencias nutricionales y genéticas. La mayoría de los casos de homocisteína elevada (dos/tercios) en la población en general se debe a deficiencias de ácido fólico, vitamina B6 y vitamina B12.
La homocisteína formada en el hígado puede seguir dos vías:
a) pasar a la sangre, ruta sistémica, y llegar a los tejidos periféricos.
b) seguir la ruta metabólica. Esta a su vez puede ser doble, ya que puede remetilarse a metionina (vía anabólica) o trans-sulfurarse, pasando a cisteína y eliminándose por la orina en forma de sulfato (vía catabólica). Entonces la homocisteína constituye la conexión entre la vía de transulfuración y la remetilación a metionina, que se realiza por dos reacciones alternativas, de las cuales la principal está implicada en el metabolismo de los folatos y la cobalamina. Ver figura debajo.
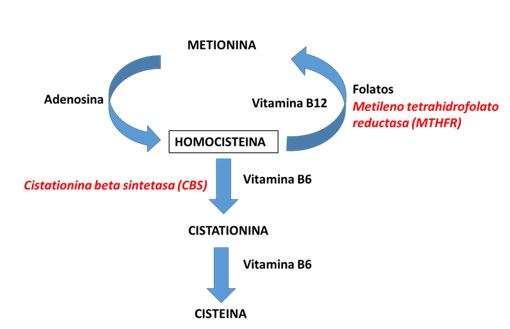
Se han descrito errores congénitos con graves consecuencias patológicas en numerosos pasos de estas vías metabólicas, siendo las más importantes la Homocistinuria, la Cistationinuria y la Deficiencia de sulfito-oxidasa. Se observan también alteraciones secundarias asociadas a otras enfermedades, como la hipermetioninemia en algunas hepatopatías graves y la cistationinuria asociada al neuroblastoma y a la deficiencia de piridoxina.
El defecto más frecuente del metabolismo de los aminoácidos sulfurados es la HOMOCISTINURIA, que puede ser causada por bloqueo enzimático a diferentes niveles, esencialmente tres: cistationina-ß-sintetasa (Homocistinuria clásica); 5,10-metilenotetrahidrofolato-reductasa; y 5-metiltetrahidrofolato-homocisteína-metiltransferasa.
La medición de la concentración de homocisteína total en sangre, es indispensable ya que incluye las diferentes formas de este aminoácido que se hallan en equilibrio: homocisteína ligada a proteínas (hasta un 70-80% de la total), disulfuro de cisteína, y homocisteína y homocisteína libre (en muy baja concentración en el individuo sano), rápidamente oxidada a homocistina.
Se sabe que la elevación plasmática de la homocisteína es el factor responsable de las principales manifestaciones clínicas, como las alteraciones vasculares y las complicaciones tromboembólicas, debido a su toxicidad sobre el endotelio de los vasos sanguíneos, mayor adherencia plaquetaria y aumento de la proliferación de las células del musculo liso.
La hiperhomocisteinemia también afecta a la síntesis del colágeno y de la elastina del tejido conjuntivo, y por ello, se observan alteraciones óseas, cutáneas, y ectopia lentis. El retraso mental que se presenta en el 50% de los pacientes afectados, parece ser debido al déficit de cistationina (aminoácido importante en la composición cerebral) y a la inhibición competitiva del transporte de aminoácidos al cerebro y formación de neurotransmisores por la elevada concentración de metionina y homocisteína.
El diagnóstico diferencial de los defectos metabólicos causantes de Homocistinuria se basa en la observación del perfil de aminoácidos en suero y orina. La cuantificación de estos se puede realizar por HPLC con detección fluorimétrica o electroquímica o bien por procedimientos inmunoenzimáticos automatizados (hay elevación en plasma y orina de homocisteína y metionina con disminución de cistina y cistationina).
Para la detección de homocisteína en orina, se utiliza el Test del Cianuro Nitroprusiato, técnica de screening para demostrar el aumento de la eliminación de los compuestos que contienen sulfhidrilo en la orina. Los sulfhidrilos oxidados son reducidos con cianuro de sodio para luego reaccionar con el nitroprusiato de sodio (con grupos sulfhidrilos libres) produciendo una coloración de rosa. Para diferenciar entre homocisteína y cistina se utiliza la Técnica del nitroprusiato de plata: la homocistina es reducida a la forma reactiva (homocisteína) por el nitrato de plata, mientras que la cistina permanece en la forma oxidada (no reactiva). La homocistina reacciona con el nitroprusiato para dar un complejo púrpura.
El ensayo enzimático para la detección de la homocisteína en sangre se basa en un novedoso principio de test “Enzyme cycling assay” (en español: ensayo cíclico enzimático) en que se determina el producto de conversión del cosubstrato y no directamente el cosubstrato o los productos de conversión de la Homocisteína.
Prestaciones disponibles en Cibic:
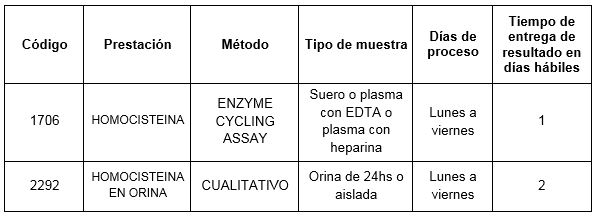
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Referencias
– Control Global del riesgo metabólico. Vol. 1 – Jose Saban Ruiz (Editor)
– Arderiu, M.J. Castiñeiras Lacambra, J.M. Queraltó Compañó Bioquímica clínica y patología molecular. Volumen II – X. Fuentes
– Couce ML1; Balcells S2; Dalmau J3; Grinberg D2; Rodés M4; Vilaseca MA5Protocolo de diagnóstico y tratamiento de homocistinuria
– Diagnostico de homocistinuria y deficiencia de Adenilosuccinato Liasa a través de técnicas químicas, bioquímicas y moleculares. Marta Bermúdez PhD. Instituto de Genética Humana Pontificia Universidad Javeriana, Bogotá, Colombia – Universidad Colegio Mayor de Cundinamarca, Bogotá, Colombia. Yina Carrillo Bsc. Universidad Colegio Mayor de Cundinamarca, Bogotá, Colombia. Recibido:13-04-2005 / Aceptado:26-05-2005
– Kaul S, Zaheh AA, Shah PK. Homocysteine hypothesis for atherothrombotic cardiovascular disease. JACC 2006;48(5):914-23.
– Mudd SH, Levy HL, Skovby F: Disorders of Transsulfuration. In: Scriver CR, Beaudet AL, Sly WS, et al., editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill, 1995:1279-1327.
– Refsum H, Smith AD, Ueland PM, et al. Facts and recommendations about total homocysteine determinations: and expert opinion. Clin Chem 2004; 50(1):3-32.
Para mayor información o consultas:
Sección: Bioquímica Clínica
Bioq. María Florencia Mora Dengra
Tel: 0341-4722424. Int: 222.