
La fibrosis quística (FQ) es una de las enfermedades hereditarias autosómicas recesivas más frecuentes en la población caucásica, con una frecuencia estimada de 1 cada 3000 nacidos vivos (1). Esta enfermedad ataca las células epiteliales exocrinas y las personas afectadas producen un moco espeso y viscoso, lo que provoca una obstrucción de los conductos de los órganos donde se localiza, siendo el páncreas y los pulmones los órganos más comprometidos.
El gen de la FQ (gen CFTR), comprende 27 exones y codifica para una proteína de 1480 aminoácidos denominada CFTR (del inglés: regulador de la conductancia transmembrana de la FQ) que se localiza en la membrana apical de las células epiteliales normales y actúa como un canal de cloro regulador del transporte iónico. Las mutaciones en el gen CFTR se distribuyen a lo largo de toda su secuencia nucleotídica, en la actualidad se han descripto más de 1500. La mutación más común a nivel mundial es una deleción de una fenilalanina en la posición 508 (p.F508del) (2).
Figura: estructura de la proteína CFTR.
Hoy en día, gracias a los avances en farmacología y genética, los niños que nacen con esta patología tienen una esperanza de vida más larga y una mejor calidad de vida que en el pasado. Durante los últimos 10 años, las investigaciones han ayudado a entender mejor esta enfermedad y a desarrollar nuevos tratamientos que nos permiten pensar que algún día se logre la cura mediante la reparación del gen involucrado ya sea mediante terapia génica o celular.
Aspectos diagnósticos
El diagnóstico de la FQ se ha basado clásicamente en por lo menos tres determinaciones positivas de electrolitos en sudor, más la presencia de al menos una de las siguientes características clínicas:
- alteraciones gastrointestinales y nutricionales
- síndrome de pérdida de sal
- azoospermia obstructiva
- antecedentes familiares
- pesquisa neonatal con resultado positivo
Desde que se conoce el gen defectuoso de la FQ, se han publicado diferentes casos de enfermos con ciertas mutaciones infrecuentes, que pueden estar asociados a una prueba del sudor normal, o expresiones clínicas atípicas, por lo que se propone que para efectuar el diagnóstico sea necesario por lo menos uno de los criterios clínicos, junto con la determinación patológica de electrolitos en sudor o bien el hallazgo de dos mutaciones en el gen CFTR (3).
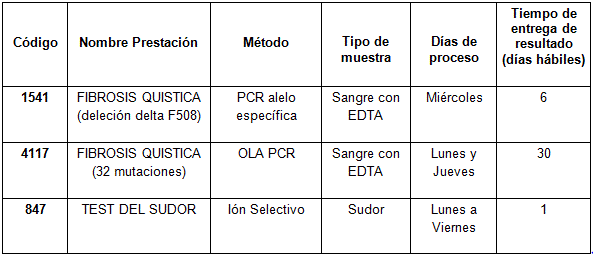
Determinaciones disponibles en CIBIC:
Bibliografía
1. Welsh MJ, et al. Cystic Fibrosis. The metabolic and molecular bases of inherited disease. Mc Graw-Hill; 2001: 5121-5188.
2. Dean M, Santis G. Heterogeneity in the severity of cystic fibrosis and the role of CFTR gene mutations. Human Genetics. 1994;93:364-368.
3. Consenso nacional de fibrosis quística. Arch Argent Pediatr 2008; (Supl) 106(5):e01-52.
Para mayor información o consultas:
Sección: Biología Molecular
Dra. María Florencia Gosso
Lic. Analía Seravalle
Tel: 0341-4499444. Internos: 242 – 258