
{kind=link}
La atrofia muscular espinal ó AME (SMA, por sus siglas en inglés) es una enfermedad neurodegenerativa caracterizada por la afectación de las células del asta anterior de la médula espinal, con debilidad proximal y simétrica y atrofia progresiva de los grupos musculares. El nivel de afectación de esas neuronas no es similar en todos los pacientes y se la clasifica generalmente sobre la base de la gravedad de los síntomas, edad de aparición y evolución en cuatro grupos: tipo I o aguda OMIM# 253300(1), tipo II o intermedia OMIM# 253550(1), tipo III o juvenil OMIM# 253400(1) y tipo IV ó del adulto OMIM# 182980(1)).
Con una incidencia aproximada de 1/6000 a 1/10000 nacimientos, y una frecuencia de portadores de 1/40 – 1/60, la AME es considerada una de las principales causas hereditarias de mortalidad infantil. Presenta un patrón de herencia autosómico recesivo(4).
El gen responsable de esta enfermedad se denomina Survival Motor Neuron 1 (SMN1, OMIM: 600354) localizado en el brazo largo del cromosoma 5 (5q13) (3,4). La ausencia del exón 7 del gen SMN1 se detecta en 95-98% de los afectados con AME, mientras que en el restante 2-5% se encuentra la deleción del exón 7 en una de las copias del gen y una mutación puntual en la copia restante (5).
La proteína SMN mantiene la sobrevida de las neuronas motoras, lo que permite el transporte axonal normal y el mantenimiento de la integridad de la unión neuromuscular (6), su alteración puede causar degeneración de las neuronas motoras del asta anterior de la medula espinal, las cuales son especialmente vulnerables a las alteraciones de esta proteína (7).
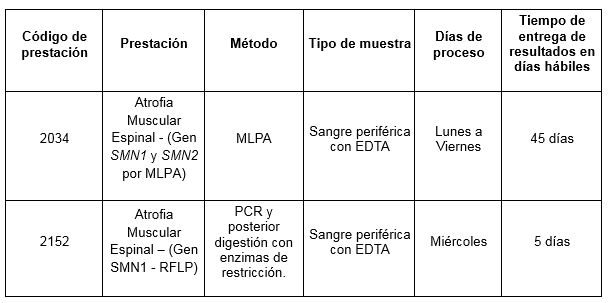
El algoritmo de diagnóstico molecular para AME comienza con el test de deleción del exón 7 (por RFLP). En el caso de obtener un resultado negativo y que la sospecha clínica persista, el paso siguiente para descartar o confirmar la enfermedad es cuantificar el número de copias del gen SMN1 (por MLPA).
Dada la ya mencionada alta frecuencia de portadores de la enfermedad, el test de cuantificación de copias del gen SMN1 también se realiza en pacientes asintomáticos con historia familiar confirmada de AME, ovodonantes y como test preconcepcional en parejas con deseos de concebir un hijo.
Prestaciones disponibles en Cibic:
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones.
Referencias
1. http://www.omim.org/
2. Van der Steege G, Grootscholten PM, van der Vlies P, Draaijers TG, Osinga J, Cobben JM, Scheffer H, Buys CH. PCR-based DNA test to confirm clinical diagnosis of autosomal recessive spinal muscular atrophy. Lancet.1995 Apr 15;345(8955):985–986.
3. Lefrbvre S, Burglen L, Reboullete S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zemani M, Le Paseur D, Fresal J, Cohen D, Weissenbach J, Munnich A, Melki J. Identification and characterization of a spinal muscular atrophy-determining gene. Cell.1995.80:155-165.
4. Tizzano EF,Cabot C,Baiget M. Cell-specific survival motor neuron gene expression during human development of the central nervous system: implications for the pathogenesis of spinal muscular atrophy. Am J Pathol.1998 Aug;153(2):355-61.
5. Prior TW, Nagan N, Sugarman EA, MS, CGC2 , Batish SD, and Braastad C. Technical standards and guidelines for spinal muscular atrophy testing. Genet Med 2011;13(7):686-694.
6. Chan YB, Miguel-Aliaga I, Franks C, Thomas N, Tru¨lzsch B, Sattelle DB, Davies KE, and van den Heuvel M. Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum Mol Gen 2003;12:1367-1376.
7. Morse R, Adrian G. Mutations in the survival motor neuron (SMN) protein alter the dynamic nature of nuclear bodies. Neuromol Med 2011;13:77-87.
Para mayor información o consultas:
Sección Biología Molecular
Lic. Analía Seravalle Tel: 0341 4499444 Int: 242
Dra. María Florencia Gosso Tel: 0341 4499444 Int: 258
Lic. Guadalupe Méjico Tel: 0341 4499444 Int: 240