
La anemia de Fanconi (FA, por su siglas en inglés) es una enfermedad hereditaria de muy baja frecuencia entre la población (1:360.000). FA se caracteriza por presentar una clínica y genética heterogéneas asociadas a inestabilidad genómica. En la actualidad, 15 genes han sido asociados a FA, siendo el gen FANCA – anemia de Fanconi, grupo complementario A – (OMIM*607139) el responsable del 60 – 70% de los casos de FA (1,3).
Las características clínicas más frecuentemente halladas (60-85%) incluyen retraso del crecimiento pre y postnatal, malformaciones renales, cardíacas y esqueléticas, facies típica, fertilidad reducida e hipogonadismo, sordera parcial, anormalidades cutáneas como hiper o hipo-pigmentación y manchas “café con leche” y elevados niveles de a-fetoproteína en sangre. La ausencia de estas características clínicas (25-40%) no es excluyente para el diagnóstico de FA.
Los problemas hematológicos aparecen, en promedio, a la edad de 7-8 años, aunque esto suele ser muy variable. Las alteraciones hematológicas afectan a un 98% de los pacientes antes de los 40 años. Estas son muy diversas, pero generalmente incluyen anemia aplásica (90%) y alto riesgo de a desarrollar diversos tipos de cáncer, principalmente leucemias de estirpe mieloide (10-30%). Además, a medida que aumenta la edad de estos pacientes, aumenta la frecuencia de tumores sólidos, fundamentalmente hepáticos y del tracto digestivo, principalmente en cabeza y cuello (25-30%).
FA posee un patrón de herencia autosómica recesiva, siendo necesaria la identificación de dos copias mutadas del gen. Dos tipos de mutaciones han sido descriptas, aquellas que originan una proteína FANCA anormal y mutaciones asociadas a la ausencia total de proteína FANCA (2). La edad de inicio de la anemia y la incidencia de leucemia es más temprana en aquellos pacientes con mutaciones asociadas a la ausencia total de proteína (3).
Determinaciones disponibles en CIBIC:
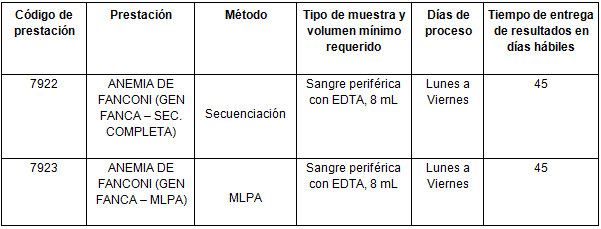
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Bibliografía
1. Apostolou S et al. Fanconi anaemia/ Breast cancer consortium. Positional cloning of the Fanconi anaemia group A gene. Nat Genet. 1996; 14:324–8.
2. Gille JJP et al. Diagnosis of Fanconi Anemia: Mutation Analysis by Multiplex Ligation-Dependent Probe Amplification and PCR-Based Sanger Sequencing. Anemia. 2012 Volume 2012, Article ID 603253.
3. Faivre L et al. Association of complementation group and mutation type with clinical outcome in Fanconi anemia. European Fanconi Anemia Research Group. Blood. 2000; 96:4064-70.
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Analía Seravalle Tel: 0341 4499444 Int: 242
Dra. María Florencia Gosso Tel: 0341 4499444 Int: 258