
El Síndrome de Rett (RTT, MIM#312750) es la causa más común de retraso mental en mujeres. RTT es una enfermedad neurogenética la cual es heredada en forma dominante ligada a X. Se caracteriza por desarrollo psicomotor normal entre los 6 – 18 meses de vida, luego de este período normal en el desarrollo, se manifiestan un retraso grave en la adquisición del lenguaje y coordinación psicomotora, así como retraso mental grave a severo.
RTT, en su variante con habla preservada (variante Zappella), está asociado a mutaciones puntuales (80%) y en menor frecuencia a duplicaciones / deleciones (8%) en el gen MECP2 (MIM*300005), localizado en el cromosoma X (Xq28). RTT afecta aproximadamente 1 en 10000 niñas nacidas vivas (1,2). Las mutaciones en el gen MECP2 suelen ser letales cuando ocurren en varones, pero se han reportado casos en donde se observa encefalopatía neonatal severa, siendo este el fenotipo más característico asociado a RTT en varones (3). En un 99% los casos, RTT es producido por mutaciones esporádicas. En la actualidad han sido reportadas más de 200 mutaciones en el gen MECP2 asociadas a RTT (4).
Determinaciones disponibles en CIBIC:
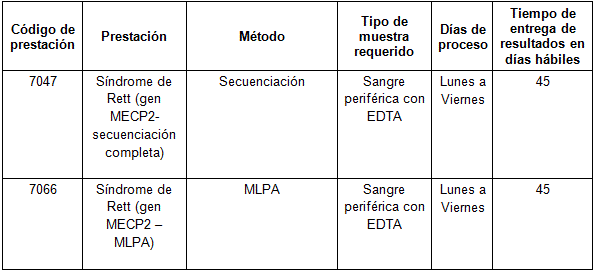
Para conocer las condiciones del paciente, de almacenamiento y de envío de las muestras y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Bibliografía
1) Neul JL y cols. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 2010; 68:944-50.
2) Scala E y cols. MECP2 deletions and genotype-phenotype correlation in Rett syndrome Am J Med Genet A. 2007; 143:2775-84.
3) Villard L, Kpebe A, Cardoso C, Chelly PJ, Tardieu PM, Fontes M. Two affected boys in a Rett syndrome family: clinical and molecular findings. Neurology. 2000; 55:1188–93.
4) RettBASE, http://mecp2.chw.edu.au
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Analía Seravalle Tel: 0341 4499444 Int: 242
Dra. María Florencia Gosso Tel: 0341 4499444 Int: 258