

El síndrome de deleción 22q11.2 es el síndrome de microdeleción más común. En un principio se pensaba que deleciones del cromosoma 22 (CATCH 22), Síndrome de DiGeorge, Síndrome Velocardiofacial y Conotruncal eran trastornos separados, pero ahora se sabe que forman parte del mismo síndrome (1).
El diagnóstico es extremadamente difícil debido a la gran variabilidad en las presentaciones fenotípicas, las cuales pueden incluir: defectos faciales, cardiopatías congénitas, hipoplasia tímica, hipoparatiroidismo, alteraciones renales, psiquiátricas e inmunológicas. Debido a esto último es que las personas afectadas presentan mayor riesgo de desarrollar infecciones y también presentan riesgo más elevado de desarrollar una enfermedad autoinmune. En algunos casos se presenta un fenotipo completo, mientras que en otros se presenta en formas muy atenuadas, reconocidas tardíamente (2).
En la mayoría de los casos, el síndrome se debe a una deleción de 3 millones de pares de bases (Mb) en la región cromosómica 22q11.2 que está flanqueada por repeticiones de bajo número de copias, producida por una recombinación meiótica no alélica durante la espermatogénesis u ovogénesis. En aprox. el 15% de los casos, la deleción es de menor tamaño, pero dentro de la región de 3 Mb, y en estos casos suele ser de tamaño variable. También hay deleciones atípicas que están ubicadas en la región crítica de DiGeorge. Algunas de ellas incluyen el gen TBX1 que está implicado en el desarrollo cardíaco, de paratiroides, del timo y de la estructura facial; y el gen HIRA (también llamado TUPLE1). Se cree que la expresión variable del fenotipo 22q11.2 se debe a modificadores genéticos en el otro alelo de 22q11.2 o en otros cromosomas (3).
Aunque la pérdida en 22q11.2 que produce los síndromes de DiGeorge y Velocardiofacial es generalmente de novo, entre un 6 a 28% de los pacientes pueden tener la alteración cromosómica heredada. En alguno de estos casos heredados se ha observado que uno de los padres puede tener la microdeleción 22q11.2 en un cromosoma del par 22, y una microduplicación 22q11.2 en el otro (por lo que la región no se pierde). En estos casos hay un incremento del riesgo de repetición en otros hijos. Por tanto, siempre se debe estudiar a los padres de niños con microdeleción con el fin de realizar un buen asesoramiento genético (4).
Cuando se sospecha la alteración, el cariotipado convencional solo es capaz de detectar un pequeño porcentaje de deleciones cromosómicas. Sin embargo, mediante hibridación fluorescente in situ (FISH) se pueden detectar muchas deleciones y microdeleciones, o bien, por biología molecular mediante MLPA (Multiplex Ligation dependent Probe Amplification) (1).
El FISH es una técnica que detecta secuencias de ADN en células o tejidos preservados mediante el empleo de una sonda (fragmento de ADN homólogo a la secuencia blanco) marcada con un fluorocromo, la cual va dirigida hacia un lugar específico del ADN y que emite fluorescencia que puede ser observada por medio de un microscopio de epifluorescencia (5).
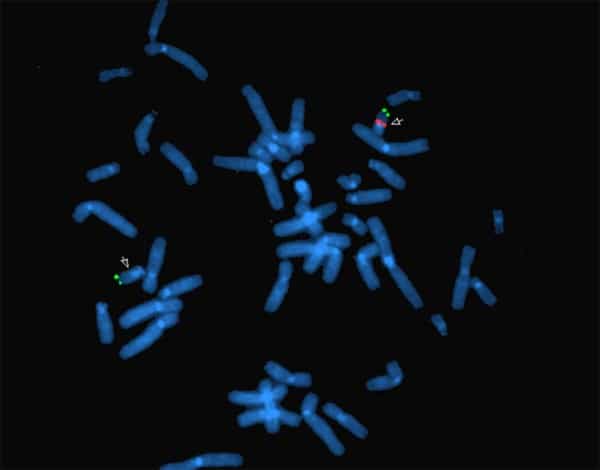
Figura 1: Delecion 22q11.2. Núcleo en metafase
Esta técnica ha demostrado ser de gran valor para la identificación de aberraciones cromosómicas clínicamente significativas, ya que el FISH se puede realizar en metafases o en células en interfase que no se dividen, y puede detectar anormalidades genómicas con una resolución de 150 a 900 kb dependiendo del tamaño de la sonda (6) (Fig. 1 y 2).
Un núcleo normal mostrará 2 señales (generalmente rojas) correspondientes a la región en estudio y 2 señales control (generalmente verdes) de una sonda que hibrida sobre el mismo cromosoma, pero en una región distante. Si el gen está delecionado o duplicado, el patrón de señales rojas esperado será distinto de 2.
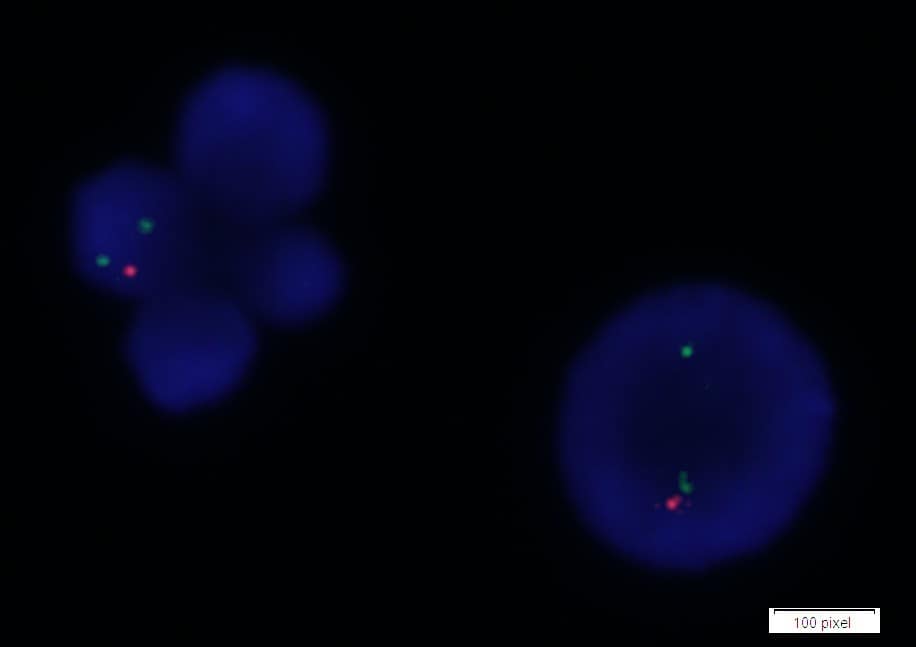
Figura 2: Deleción 22q11.2: Núcleos en interfase.
Debido a que se presume que dos de los principales genes responsables de la mayoría de las malformaciones físicas serían el gen TBX1 y el HIRA (también llamado TUPLE1), Cibic Laboratorios ofrece para una primera exploración la sonda de LIVe MD22q11.2 que hibrida sobre los loci TBX1 y Tuple1 (o HIRA) y contiene además un control de reacción sobre el locus ARSA. Por otra parte, también disponemos de la técnica de MLPA, de mayor sensibilidad y tiempo de proceso, para explorar con mayor detalle posibles deleciones en estas regiones.
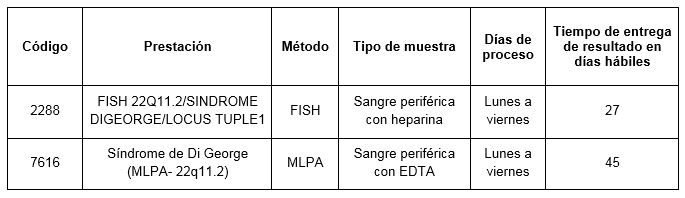
Prestaciones disponibles en Cibic Laboratorios:
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Referencias:
1. Kimberley A. Miller, MSN, RNC, NNP (2008). FISH Diagnosis of 22q11.2 Deletion Syndrome
2. Lucía Sierra Santos, Pilar Casaseca García, Alfonso García Moreno y Vicente Martín Gutiérrez (2014). Síndrome de Di George
3. El síndrome de deleción 22q11.2 – Enciclopedia Orphanet de la Discapacidad, octubre de 2017
4. María Luisa Martínez-Fernández, Alexandra MacDonald, Isabel Aceña, María Dolores SánchezIzquierdo, Eva Bermejo y María Luisa Martínez-Frías. Madrid 2010. Síndromes de DiGeorge, Velocardiofacial y Microdeleción 22q11.2. Edita: Estudio Colaborativo Español de Malformaciones Congenitas.
5. Gersen SL, Keagle MB. (2013). The Principles of Clinical Cytogenetics. USA. Humana Press.
6. Arsham MS, Barch, MJ Lawce HJ. (2017). The AGT Cytogenetics Laboratory Manual. 4th Edition. Wiley-Blackwell.
Para mayor información o consultas:
Sección: Citogenética
 Bioq. Sibila Bertalot
Bioq. Sibila Bertalot
 Téc. Verónica Vanrell
Téc. Verónica Vanrell
Tel: 0341-472 2424. Interno: 283
Sección: Biología Molecular
 Lic. Analía Seravalle
Lic. Analía Seravalle
Tel: 0341-4722424. Interno: 242.
 Lic. Alan Gómez
Lic. Alan Gómez
Tel: 0341-4722424. Interno: 225.