

Las enfermedades cardiovasculares (ECV) conforman una de las mayores causas a nivel mundial de enfermedad y mortalidad, afectando a más de 16 millones de individuos anualmente. Tradicionalmente, el foco de atención ha sido puesto en lo que suelen denominarse factores de riesgo individuales asociados al estilo de vida (ej. dieta, tabaquismo, ejercicio) dado que las ECV se hallan frecuentemente relacionada a su vez, con otras enfermedades tales como diabetes y obesidad. Sin embargo, durante los últimos años, con el fin de diagnosticarlas en forma inequívoca, se ha prestado especial atención al estudio e identificación de factores de riesgo genéticos.
La Displasia/ Miocardiopatía arritmogénica del ventrículo derecho (D/MAVD): es una enfermedad del músculo cardíaco caracterizada por anormalidades estructurales y funcionales del ventrículo derecho principalmente, con sustitución progresiva del miocardio por tejido graso y fibroso tras un inadecuado proceso apoptótico. Afecta a seis de cada 10.000 individuos. Las manifestaciones clínicas de D/MAVD abarcan individuos asintomáticos, arritmias ventriculares e insuficiencia cardíaca derecha o biventricular (1). D/MAVD constituye una de las principales causas de muerte súbita en adultos jóvenes con una incidencia aún mayor entre los deportistas (2).
La agregación familiar se demuestra entre un 30-50% de los casos (3). D/MAVD es heredada con un patrón de herencia autosómico dominante con penetrancia y expresividad variable. También han sido reportadas formas autosómicas recesivas de la enfermedad (ej. Síndrome de Naxos y Síndrome de Carvajal) (4). El mecanismo patológico de D/MAVD se presume es debido a una adhesión anormal por parte de los miocitos, abarcando alteraciones a nivel de las uniones intercelulares (5). A nivel de las uniones intercelulares, los desmosomas cumplen una función esencial en mantener la unión de los filamentos intermedios asociados al citoesqueleto. Mutaciones en varios genes que codifican proteínas asociadas a la compleja estructura de los desmosomas han sido asociadas a D/MAVD, siendo las principales PKP2, DSP, DSG2, DSC2 y JUP (ver tabla).
Es fundamental para este tipo de patologías el correcto diagnóstico molecular en el menor tiempo posible, impactando esto directamente en la toma de decisiones por parte de los médicos especialistas.
Las dificultades en el diagnóstico de mutaciones asociadas a CMH por técnicas de secuenciación tradicionales (tipo Sanger), radican en la elevada demanda en tiempo de mano de obra especializada debido al elevado número de genes que deben ser analizados. En la actualidad, es posible la realización de este tipo de estudios en forma paralela (ej. sobre la misma muestra, secuenciación simultánea de todos los genes sospechados) mediante secuenciación de segunda generación NGS (Next generation Sequencing, por sus siglas en Inglés).
Lo anteriormente mencionado impacta en forma directa en:
(i) el tiempo que demora la obtención de un correcto diagnóstico molecular siendo crítico el tiempo efectivo en el que un resultado es entregado al especialista solicitante.
(ii) costo-efectividad de el/los análisis a realizar (ej. la secuenciación de un único gen puede llevar varias semanas de trabajo, aumentando este tiempo si más de un gen debe incluirse en el pipeline del diagnóstico molecular).
Prestación disponible en Cibic:
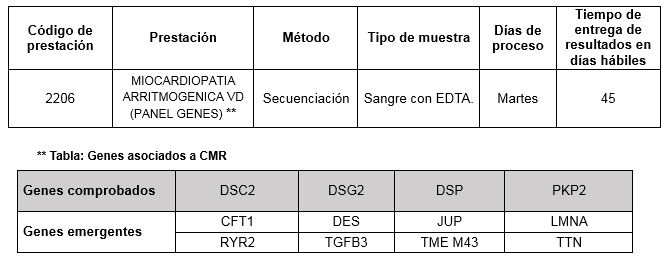
Los genes comprobados son aquellos en los cuales se han descripto mutaciones como causantes de la patología en estudio.
Los genes emergentes son genes en los cuales mutaciones asociadas a la patología en estudio han sido asociados recientemente.
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Referencias
1- Basso C y cols. Arrhythmogenic right ventricular cardiomyopathy. Lancet 2009; 373: 1289-1300.
2- Thiene G y cols. Right ventricular cardiomyopathy and sudden death in young people. N Engl J Med 1988; 318: 129-133.
3- James CA y cols. Update on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C). Curr Treat Options Cardiovasc Med 2013; 15: 476-487.
4- Protonotarios N y cols. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol 2004; 13:185-194.
5- Fressart V, Duthoit G y cols. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia
Para mayor información o consultas:
Sección: Biología Molecular
Dra. María Florencia Gosso.Tel.: 0341-4499444. Int: 258.

Lic. Guadalupe Méjico. Tel.: 0341-4499444. Int: 239.

Lic. Analía Seravalle.Tel.: 0341-4499444. Int: 242.
