
La deficiencia de alfa-1-antitripsina (A1AT) es un desorden hereditario causado por mutaciones en el gen SERPINA 1 que puede ocasionar entre la tercera y cuarta década de vida una enfermedad pulmonar obstructiva crónica (EPOC), como enfisema y bronquitis crónica (1, 2). Su comienzo puede acelerarse en el caso de fumadores y sujetos expuestos a severa polución ambiental. Con menor frecuencia se puede manifestar desde el nacimiento hasta cualquier momento en la vida como una enfermedad hepática crónica. La A1AT es un inhibidor de serin proteasas producida por el hígado cuya principal función es proteger al pulmón contra el daño proteolítico de la elastasa secretada por los neutrófilos en respuesta a infecciones e irritantes inhalados, como el humo del cigarrillo.
La deficiencia de A1AT se caracteriza por bajos niveles circulantes de la proteína A1AT, lo cual reduce la protección del tejido pulmonar contra la actividad elastasa, conduciendo a la destrucción del tejido y finalmente al desarrollo temprano de EPOC (3). Las enfermedades hepáticas, incluyendo hepatitis, cirrosis y hepatoma son causadas por la proteína A1AT mutada que se acumula en los hepatocitos (2, 3). El gen SERPINA 1 es altamente polimórfico y está organizado en 4 exones codificantes (2, 3, 4 y 5) y 3 exones no codificantes (1a, 1b, y 1c). Se conocen al menos 100 alelos diferentes pero muchos de estos no afectan la expresión o función de A1AT. Sin embargo hay variantes que están asociadas con una menor concentración de A1AT o con la producción de una proteína no funcional. Estos alelos asociados con enfermedad se pueden clasificar de acuerdo a su mecanismo patogénico: alelos deficientes, conducen a la acumulación intracelular o degradación de A1AT resultando en cantidades deficientes de proteína en circulación; alelos nulos, no producen transcripto, o producen una proteína truncada o una proteína inestable, que en definitiva es completamente degradada antes de ser secretada; y alelos disfuncionales, que son aquellos que codifican para una proteína con menor actividad inhibitoria de la elastasa (4-6).
Los alelos más comunes en la población general son el M, el S y el Z. El alelo M es considerada la variante normal y se asocia con niveles normales de A1AT en plasma. Las variantes más comunes asociadas a enfermedad son el alelo deficiente S (E288V), asociado con niveles reducidos de A1AT; y el alelo Z (E366K) que codifica para una proteína inestable que se acumula dentro del hepatocito y que a su vez posee menor actividad enzimática. Los pacientes homocigotas para el alelo S tienen concentraciones de A1AT entre un 10 a 20% menores pero que son suficientes para prevenir las manifestaciones clínicas de deficiencia de A1AT. Sin embargo, en presencia de un alelo Z (genotipo SZ), las concentraciones de A1AT son menores y podría conducir a una actividad elastasa descontrolada aumentando el riesgo de desarrollar enfermedad pulmonar obstructiva crónica (pero no enfermedad hepática). Por otro lado, los pacientes homocigotas para el alelo Z (genotipo ZZ) tiene un alto riesgo de enfermedad hepática de comienzo temprano y enfisema prematuro (6-10).
Si bien la deficiencia de A1AT es uno de los desórdenes hereditarios más prevalentes y potencialmente letal, está sub y mal diagnosticado, puede ser malinterpretado como asma y rara vez se diagnostica antes de que se desarrollen los síntomas clínicos tales como la enfermedad pulmonar obstructiva crónica. El retardo en el diagnóstico también retarda oportunidades de prevención y terapia (11).
Se recomienda considerar el diagnóstico en:
– Evaluación inicial de todo paciente con EPOC.
– Pacientes con asma de control incompleto luego de un tratamiento agresivo con broncodilatadores
– Pacientes con bronquiectasias (dilataciones de los bronquios)
– Recién nacidos, niños y los adultos con una enfermedad hepática inexplicable (hepatitis neonatal, colestasis neonatal, cirrosis y cáncer del hígado)
– Personas con antecedentes familiares de enfermedades hepáticas
– Familiares o parientes consanguíneos de una persona diagnosticada con deficiencia de A1AT.
La determinación de los niveles de A1AT es un test simple y ampliamente disponible. Sin embargo, no siempre es útil ya que como es una proteína de fase aguda, puede aumentar su concentración sérica a niveles normales durante procesos inflamatorios o trauma. La genotipificación de los alelos S y Z simplifica el diagnóstico de deficiencia de A1AT y debe interpretarse junto con el dato de la concentración sérica de A1AT. El análisis fenotípico por isoelectroenfoque o la secuenciación completa del gen SERPINA 1 son necesarios para clarificar resultados discordantes entre los datos del genotipo y niveles bajos de proteína sérica (12).
Determinaciones disponibles en Cibic:
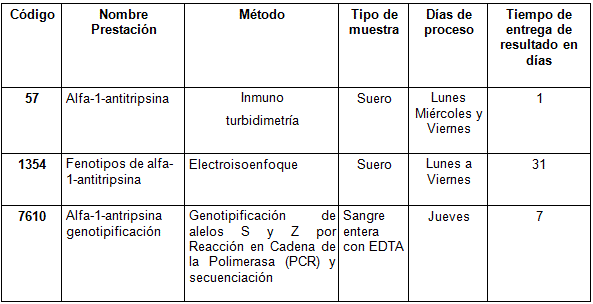
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y la extranet.
Referencias
1- Stoller JK, Aboussouan LS. a1-Antitrypsin deficiency. Lancet 365:2225–2236, 2005.
2- Kohnlein T and Welte T. Alpha-1 antitrypsin deficiency: pathogenesis, clinical presentation, diagnosis, and treatment. Am J Med; 121: 3-9, 2008.
3- Crystal RG. Alpha 1-antitrypsin deficiency, emphysema, and liver disease. Genetic basis and strategies for therapy. J Clin Investigation 85:1343-1352, 1990.
4- Wood AM and Stockley RA. Alpha one antitrypsin deficiency: from gene to treatment. Respiration 74: 481-492, 2007.
5- Demeo DL, Silverman EK. alpha1-Antitrypsin deficiency. 2: genetic aspects of alpha1-antitrypsin deficiency: phenotypes and genetic modifiers of emphysema risk. Thorax 59:259–264, 2004.
6- Lomas DA, Parfrey H. alpha1-Antitrypsin deficiency. 4: molecular pathophysiology. Thorax 59: 529–35, 2004.
7- Fregonese L, Stolk J, Frants RR and Veldhuisen B. Alpha-1 antitrypsin Null mutations and severity of emphysema. Respiratory Med 102: 876-884, 2008.
8- Lomas DA, Mahadeva R. alpha1-Antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest 110: 1585–1590, 2002.
9- Perlmutter DH. Liver injury in alpha1-antitrypsin deficiency: an aggregated protein induces mitochondrial injury. J Clin Invest 110:1579–1583, 2002.
10- Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z a1-antitrypsin accumulation in the liver. Nature 357:605–607, 1992.
11- Brantly M. Efficient and accurate approaches to the laboratory diagnosis of #1-antitrypsin deficiency: the promise of early diagnosis and intervention. Clin Chem 52:2180 –2181, 2006.
12-Strange C, Dickson R, Carter C, Carpenter MJ, Holladay B, Lundquist R, Brantly ML. Geneticm testing for alpha1-antitrypsin deficiency. Genet Med 6:204-210, 2004.
Para mayor información o consultas:
Sección: Biología Molecular
Dra. María Sonia Baquedano
Lic. Analía Seravalle
Tel: 0341-4499444. Interno: 241 – 242