

La atrofia muscular espinal ó AME (SMA, por sus siglas en inglés) es una enfermedad neurodegenerativa caracterizada por la afectación de las células del asta anterior de la médula espinal, con debilidad proximal y simétrica y atrofia progresiva de los grupos musculares. El nivel de afectación de esas neuronas no es similar en todos los pacientes y se clasifica generalmente en cuatro grupos (tipo I o aguda OMIM# 253300(1), tipo II o intermedia OMIM# 253550 (1), tipo III o juvenil OMIM# 253400 (1) y tipo IV ó del adulto OMIM# 182980 (1) sobre la base de la gravedad de los síntomas, la edad de aparición y la evolución.
![]()
![]()
Los bebés con atrofia muscular espinal tipo I nacen con muy poco tono muscular, músculos débiles y con problemas respiratorios y de alimentación. En la atrofia muscular espinal tipo II los síntomas pueden no aparecer hasta la edad de 6 meses a dos años. La atrofia muscular espinal tipo III es una enfermedad más leve que empieza en la niñez o la adolescencia y empeora lentamente. La atrofia muscular espinal tipo IV es incluso más leve, con debilidad que comienza en la adultez.
Con una incidencia aproximada de 1/6000 a 1/10000 nacimientos, y una frecuencia de portadores de 1/40-1/60, la AME es considerada una de las principales causas hereditarias de mortalidad infantil. Presenta un patrón de herencia autosómico recesivo (2,3).
El gen responsable de esta enfermedad se denomina Survival Motor Neuron 1 (SMN1, OMIM: 600354) localizado en el brazo largo del cromosoma 5 (5q13).La patología molecular del gen SMN1 observada en los afectados incluye mayoritariamente la ausencia total del gen, deleciones y conversiones génicas, habiéndose descrito también mutaciones puntuales. Existe un gen homólogo al SMN1, el SMN2 que está presente en todos los pacientes y que expresa fundamentalmente una RNA mensajero y proteína incompletos (conocido como Δ7 dado que le falta el exón 7). El gen SMN1 está delecionado o interrumpido en más del 95% de los pacientes cualquiera que sea la forma clínica.
En aquellos casos sin deleción homocigota se ha detectado una mutación recurrente de cuatro pares de bases (delAGAG) en el exón 3 presente en el 3% de los afectados. Existen también otras mutaciones puntuales descriptas a lo largo de todo el gen.
La ausencia del exón 7 del gen SMN1 confirma el diagnóstico de AME. Aproximadamente el 90% tienen también deleción del exón 8 mientras que un 5% tiene presencia de este exón (4).
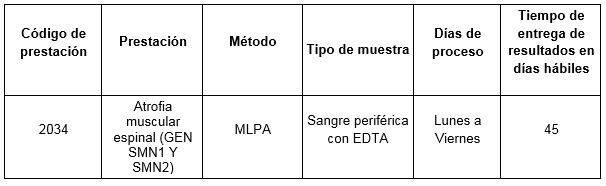
Prestación disponible en Cibic:
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte a la intranet o al manual de prestaciones.
Referencias
1. http://www.omim.org/
2. Lefrbvre S y cols. Identification and characterization of a spinal muscular atrophy-determining gene. Cell.1995.80:155-165.
3. Tizzano EF y cols. Cell-specific survival motor neuron gene expression during human development of the central nervous system: implications for the pathogenesis of spinal muscular atrophy. Am J Pathol.1998;153:355-61.
4. van der Steege G, Grootscholten PM, van der Vlies P, Draaijers TG, Osinga J, Cobben JM, Scheffer H, Buys CH. PCR-based DNA test to confirm clinical diagnosis of autosomal recessive spinal muscular atrophy. Lancet.1995;345:985–986.
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Analía Seravalle
Lic. Guadalupe Méjico
Bioq. María Florencia Gosso (PhD)
Tel: 0341 4499444 Int: 242, 243, 258