
Las distrofinopatías incluyen un espectro amplio de enfermedades neuromusculares causadas por mutaciones en el gen DMD (MIM:*300377) localizado en el cromosoma X (Xp21.2). La clínica de las distrofinopatías varía desde fenotipos asintomáticos que cursan con incremento sérico de creatina fosfoquinasa y calambres musculares asociado a hemoglobinuria, hasta fenotipos severos de enfermedad muscular progresiva los cuales son clasificados como (i) distrofias musculares de Duchenne (DMD, MIM:#310200) y de Becker (BMD; MIM#300376) las cuales afectan principalmente músculo esquelético y (ii) cardiomiopatía dilatada asociada a DMD (MIM#302045), en la cual el tejido cardíaco es el tejido principalmente afectado (1, 2).La DMD es más frecuente, temprana y grave que la DMB. La incidencia de la DMD es de 1 por cada 3.300 nacimientos de varones. La incidencia de la DMB varía entre 1 por cada 18.000 – 31.000 nacimientos de varones.
El gen DMD, localizado en el cromosoma X (Xp21.2) es el gen humano más grande conocido, y codifica una proteína llamada distrofina, que se expresa en las células musculares. Las mutaciones responsables de DMD conllevan una ausencia total de distrofina, mientras que las que subyacen a la DMB conducen a una cantidad y/o calidad anormal de la proteína (3). En ambos casos es frecuente encontrar deleciones del gen completo o de varios exones (60-65%) y duplicaciones de uno o más exones (5-10%). En menor frecuencia, puede hallarse mutaciones puntuales (20-35%).
Las distrofinopatías se heredan de forma ligada al X. Las mujeres portadoras tienen un riesgo del 50% de transmitir la mutación a su descendencia. Los hijos varones que hereden la mutación estarán afectados; las hijas que hereden la mutación serán portadoras, y poseen un elevado riesgo de desarrollar cardiomiopatía dilatada asociada a DMD (3).
Determinaciones disponibles en Cibic:
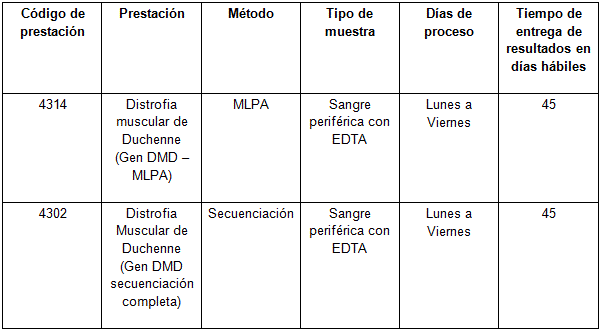
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones.
Referencias
– Chelly J y cols. Effect of dystrophin gene deletions on mRNA levels and processing in Duchenne and Becker muscular dystrophies. Cell. 1990; 63:1239–48.
– Hoffman EP y cols. The protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51: 919-28.
– Dooley J y cols. Duchenne muscular dystrophy: a 30-year population-based incidence study. Clin Pediatr (Phila). 2010. 49:177–9
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Analía Seravalle Tel: 0341 4499444 Int: 242
Dra. María Florencia Gosso Tel: 0341 4499444 Int: 258