
La Enfermedad de Von Hippel-Lindau (EVHL) es un trastorno neoplásico hereditario autosómico dominante, caracterizado por la predisposición a desarrollar tumores en numerosas y disímiles estructuras del organismo: región ocular (angiomas retinarianos), Sistema Nervioso Central (hemangioblastomas), tumor del saco endolinfático, riñones (carcinomas de células renales), glándulas suprarrenales (feocromocitomomas), páncreas (quistes pancreáticos), hígado y epidídimo. Se caracteriza por una variabilidad fenotípica – en ocasiones muy marcada – dentro de la misma familia y sus manifestaciones son tan variadas que el diagnóstico suele retrasarse muchos años (1). La prevalencia de la enfermedad es de 3 cada 100.000 individuos. Típicamente, los síntomas aparecen de la segunda a la cuarta década de la vida, con una penetrancia de la enfermedad estimada en 97% a los 60 años de edad (1). EVHL es causada por una alteración en una de las dos copias del gen oncosupresor VHL el cual se encuentra en el brazo corto del cromosoma 3, en la posición 3p25-p26. Es un gen pequeño constituído por 852 nucleótidos y 3 exones. El análisis molecular del mismo juega un rol fundamental para comprender las bases moleculares y la patogénesis de esta enfermedad. Una vez encontrada una mutación en una persona afectada, la extensión del estudio a los familiares es de suma importancia ya que permitirá identificar a los portadores de dicha mutación y realizar el seguimiento de la enfermedad de manera de poder detectar los tumores en forma precoz, como también descartar a aquellos que no han heredado la mutación y que por lo tanto no precisan ser incluidos en el protocolo de seguimiento (2). El test genético realizado en pacientes sin antecedentes familiares y con sospecha clínica de la enfermedad ha permitido identificar que un 20% de los casos se debe a mutaciones de novo, y por lo tanto representan el primer miembro afectado de la familia. Hay descriptas más de 500 mutaciones en pacientes con EVHL, que incluyen alteraciones de cambio de aminoácido, cambios que generan codones de stop, pequeñas deleciones e inserciones, y grandes reordenamientos del gen (2, 3).
La disponibilidad del estudio molecular, además de proporcionar la posibilidad de detectar tempranamente a los portadores, permite avanzar con la investigación y el desarrollo de terapias curativas. Mientras tanto, es crucial la detección precoz de los tumores y su adecuado tratamiento.
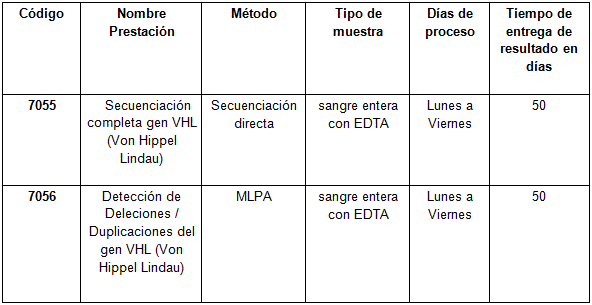
Determinaciones disponibles en CIBIC:
Bibliografía
1- Rameen Beroukhim et. al. Patterns of gene expression and copy-number alterations in VHL disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009 June 1; 69(11): 4674–4681.
2- Rolando A. Hernández Fernández. Fundamentos moleculares de la enfermedad de von Hippel Lindau. Revista Cubana de Investigaciones Biomédicas.2010; 29(2)262-273.
3- Sáenz M. P. Bases moleculares de la enfermedad de von Hippel-Lindau. Univ. Méd. Bogotá (Colombia), 49 (3): 408-412, julio-septiembre de 2008.
Para mayor información o consultas:
Sección: Biología Molecular
Dra. María Florencia Gosso. Interno: 242
Lic. Analía Seravalle. Interno: 258
Tel: 0341-4499444.