
La enfermedad de Wilson (WD, MIM#277900) se produce debido a una alteración en el metabolismo del cobre. WD puede afectar diversos sistemas del organismo, pudiéndose observar signos y síntomas a nivel hepático (ictericia recurrente, enfermedad hepática autolimitada, hepatitis de origen autoinmune, falla hepática fulminante y enfermedad hepática crónica), neurológico (distonía rígida, tremor, falta de coordinación, pérdida de control motriz fino, corea) y psiquiátricos (depresión, trastornos en la personalidad) (1).
En la actualidad, se han reportado más de 500 mutaciones asociadas al gen ATP7B (MIM*606882), el cual codifica para una proteína ATPasa del tipo P transportadora de cobre, encargada de la distribución de cobre desde el hígado hacia otros órganos y tejidos (2). Mutaciones en esta proteína originan una proteína con actividad defectuosa, con la subsecuente acumulación de cobre en el organismo, generándose depósitos de cobre en particular en hígado y cerebro. WD presenta un patrón de herencia autosómica recesiva (dos copias anormales del gen), por lo que el diagnóstico final se realiza mediante la identificación de dos mutaciones en el gen ATP7B. La prevalencia de esta patología está estimada en alrededor de 1 en 30000, con una frecuencia de portadores (solo una copia mutada del gen) de 1 en 90 (3). El grupo etario dentro del cual pueden observarse signos y síntomas es muy amplio, desde los tres años de edad hasta pasada la quinta década de vida (1).
La mayoría de los pacientes con WD poseen niveles subnormales de cobre sérico el cual es generalmente proporcional a la concentración sérica de ceruloplasmina. Esta enfermedad es sospechada en aquellos individuos que presentan niveles bajos de cobre en sangre junto con elevados niveles de cobre en orina. Si bien los niveles plasmáticos de ceruloplasmina tienden a ser bajos, este parámetro no debe ser tomado como índice excluyente ante el hallazgo de valores normales, ya que se han reportado valores normales de ceruloplasmina sérica en por lo menos 5% de casos con WD asociada a síntomas neurológicos, y en más del 40% de casos asociados a alteraciones hepáticas (4). En aproximadamente el 50-60% de los casos, debido a una alta acumulación de cobre en el organismo, es posible observar depósitos de cobre en los bordes del iris, comúnmente conocidos como anillos de Kayser-Fleischer.
Es importante tener en cuenta que los valores de cobre sérico en recién nacidos sanos son normalmente bajos. Las concentraciones de cobre en pacientes con WD se ven incrementadas durante los primeros seis meses de vida y a partir de los dos a tres años de vida, esta concentración excede ampliamente el rango normal de referencia en adultos (1). Si bien la concentración de cobre hepática en la enfermedad de Wilson suele ser mayor a 250 µg/gr de peso seco, los valores normales no sobrepasan los 55 µg/gr de peso seco (5). También pueden observarse estos niveles elevados de cobre en otras enfermedades hepáticas crónicas y en enfermedades de naturaleza colestática. Más importante aún, la concentración de cobre hepática en estadios avanzados de la enfermedad, tienden a distribuirse en forma no uniforme.
Es necesario destacar, además, que no existe correlación entre genotipo y fenotipo. WD es una patología muy heterogénea, pudiéndose observar diferencias en edad de presentación, severidad y preponderancia de síntomas hepáticos y/o neurológicos aún en pacientes que son portadores de las mismas mutaciones (1).
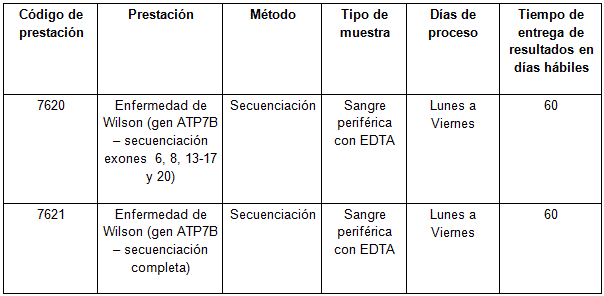
Determinaciones disponibles en Cibic:
Referencias
1. De Bie, et al. Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes. J Med Genet. 2007; 44:673-88.
2. Steindl P, et al. Wilson’s disease in patients presenting with liver disease: a diagnostic challenge. Gastroenterology. 1997;113:212-8.
3. Nuttal KL, et al. Reference limits for copper and iron in liver biopsies. Ann Clin Lab Sci. 2003;33:443-50.
4. Kenney SM y Cox DW. Sequence variation database for the Wilson disease copper transporter, ATP7B. Hum Mutat. 2007;28:1171-7.
5. Olivarez L, et al. Estimate of the frequency of Wilson’s disease in the US Caucasian population: a mutation analysis approach. Ann Hum Genet. 2001 Sep;65 (Pt 5):459-63.
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Analía Seravalle Tel: 0341 4499444 Int: 242
Dra. María Florencia Gosso Tel: 0341 4499444 Int: 258