
La fiebre mediterránea familiar (FMF) es una enfermedad hereditaria transmitida de forma autosómica recesiva, que se caracteriza por episodios recurrentes y breves de fiebre y dolor por inflamación de una o varias serosas (peritoneo, pleura, pericardio, membrana sinovial). La amiloidosis es su complicación más importante y suele ser la principal causa de muerte en los casos en que se presenta (1). Es frecuente en descendientes de poblaciones mediterráneas de Israel, Turquía, Armenia, países árabes y norte de África y menos común en griegos e italianos. La frecuencia de portadores se ha calculado en 1:5 a 1:7 en algunas de estas poblaciones, llegando la frecuencia de enfermos (homocigotos) a 1/200 individuos. (2, 3).
El inicio de los síntomas es, generalmente, en las primeras dos décadas de la vida y es excepcional que comiencen luego de los 30 años. Los ataques, pueden estar separados por días o años, y pueden ser desencadenados por sucesos ambientales, como, por ejemplo, el estrés y la actividad física extrema. Los hechos más frecuentes de estas crisis inflamatorias son: 1) fiebre recurrente, presente en todas las crisis; 2) episodios de dolor abdominal agudo en el 90% de los casos; 3) otras manifestaciones de serositis: pleuritis y pericarditis; 4) artritis, en la mayoría de los casos se trata de una oligoartritis aguda, asimétrica, que se resuelve espontáneamente. Otras manifestaciones más raras incluyen: mialgia febril, eritema tipo erisipela, vasculitis: púrpura de Henoch-Schönlein y poliarteritis nodosa, meningitis recidivante de Mollaret, esplenomegalia, orquitis, etc. (1).
La FMF está producida por mutaciones en el gen MEFV, situado en el brazo corto del cromosoma 16 (16p13), que consta de diez exones. Este gen codifica para una proteína llamada Pirina por el International FMF Consortium y Marenostrin por el French FMF Consortium (4, 5) y se expresa exclusivamente en granulocitos maduros y monocitos. No se conoce su función específica. La evidencia indica que participa en la regulación de la respuesta inflamatoria desactivando la respuesta inmune. Se ha encontrado un aumento de la expresión de los genes de varias citoquinas en los pacientes con FMF, lo que podría considerarse un estado de preactivación de células inflamatorias, por lo que se propone la persistencia de un estado de inflamación subclínica durante la remisión (6). Se han identificado más de 90 mutaciones, siendo las más frecuentes: M694V, V726A, M680I, M694I (exón 10) y E148Q (exón 2). Sin embargo, se desconoce el mecanismo exacto por el que las mutaciones en MEFV resultan en una alteración de las repuestas inflamatorias. Se ha propuesto que regula la respuesta inflamatoria a nivel del citoesqueleto leucocítico, lo que explicaría el efecto terapéutico de la colchicina (6). La mutación M694V muestra una asociación significativa, tanto en homo como en heterocigosis, con el desarrollo de amiloidosis renal. La mutación compuesta V726A-E148Q también implica una evolución más severa con mayor tendencia a la amiloidosis renal (7).
No existen marcadores específicos de enfermedad y los hallazgos de laboratorio ponen de manifiesto la existencia de un proceso inflamatorio sistémico. El estudio del gen MEFV permite establecer un diagnóstico molecular de la FMF que confirme la sospecha clínica. El diagnóstico se realiza detectando las mutaciones más frecuentes según el origen étnico del paciente o por secuenciación directa de los exones (8).
El conocimiento de las mutaciones causales tiene, además, importancia pronóstica y como guía del tratamiento ya que los portadores de determinadas mutaciones son los que tienen mayor riesgo de sufrir amiloidosis renal.
La incidencia de esta afección en nuestra población, que cuenta con un importante contingente inmigratorio de origen mediterráneo, no debe ser desestimada. Se estima que en Argentina viven 180000 descendientes de armenios y la población judía en la Argentina es la más grande de América Latina y la sexta más grande del mundo fuera de Israel. Se trata seguramente de una enfermedad subdiagnosticada, ya que si no se tiene en cuenta la genética de poblaciones que explica la aparición de la misma y no existen antecedentes familiares sugestivos, es difícil que se sospeche (2, 3).
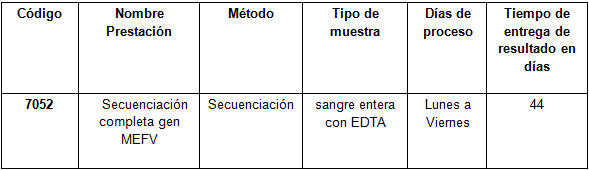
Determinación disponible en CIBIC:
Bibliografía
1. Shohat M and Halpern GJ. 2011 Familial Mediterranean fever. A review. Genet Med 13:487-498.
2. Ben-Chetrit E and Touitou I. 2009 Familial Mediterranean fever in the world. Arthritis Rheum 61:1447-1453.
3. Yepiskoposyan L and Harutyunyan A. 2007 Population genetics of familial Mediterranean fever: a review. E J Hum Genet 15.911-916.
4. The French FMF Consortium. 1997 A candidate gene for familial Mediterranean fever. Nat Genet 17: 25-31.
5. The international FMF Consortium. 1997 Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 90: 797-807.
6. Chae JJ, Aksentijevich I and Kastner DL. 2009 Advances in the understanding of familial Mediterranean fever and possibilities for targeted therapy. Br J Haematol 146: 467-478.
7. Gershoni-Baruch R, Brik R, Shinawi M, Livneh A. 2002 The differential contribution of MEFV mutant alleles to the clinical profile of familial Mediterranean fever. Eur J Hum Genet 10: 145-149.
8. Grateau G, Pecheux C, Cazeneuve C, Cattan D, Dervichian M, Goossens M. 2000. Clinical versus genetic diagnosis of familial Mediterranean fever. Q J M 93: 223-229.
Para mayor información o consultas:
Biología Molecular
Dra. María Sonia Baquedano.
Lic. Analía Seravalle.
Tel: 0341-4499444. Interno: 241 – 242