
El factor V forma parte de las proteínas que intervienen en el sistema de coagulación, ocasionalmente llamado Proacelerina o Factor Lábil. En contraste con la mayoría de los demás factores de la coagulación, el factor V no es enzimáticamente activo sino que funciona como cofactor. El gen que codifica al factor V se encuentra en el brazo corto del primer cromosoma humano (1q23). Está genómicamente relacionado con la familia de las oxidasas multicobre, y presenta una secuencia homóloga en 40% a la del factor VIII.
La glucoproteína resultante de su expresión presenta un peso molecular de aproximadamente 330 kDalton. Se sintetiza en el hígado y por los histiocitos del sistema retículoendotelial.
El factor V circula en el plasma sanguíneo como una molécula de cadena única con una vida media plasmática de aproximadamente 12 h. El factor V es capaz de unirse a las plaquetas activadas y se activa por contacto con la trombina. Al activarse, el factor V se empalma en dos cadenas que se unen de manera no-covalente entre sí por intermediación del calcio. Una vez activado el factor V cumple el rol de cofactor del factor X activo, en el complejo protrombinasa. El factor Xa requiere de calcio y del factor Va para convertir la protrombina en trombina en la superficie de la membrana celular. El factor Va es degradado por la proteína C activada, uno de los principales inhibidores fisiológicos de la coagulación (1).
El déficit congénito del factor V, llamado enfermedad de Owren o parahemofilia, forma parte de los trastornos poco comunes de la coagulación (2). Es una enfermedad hereditaria autosómica recesiva, en donde hombres y mujeres se ven afectados por igual. Los pacientes heterocigotas aunque muestran niveles bajos de factor V en plasma, probablemente nunca experimenten episodios de sangrado importante. Sin embargo los enfermos homocigotas, cuya prevalencia se estima en 1 caso por millón de habitantes, pueden presentar clínica de metrorragias, epistaxis, sangrado cutáneo o sangrado prolongado ante traumatismos o cirugías. Puede aparecer a cualquier edad, pero las formas más graves se manifiestan durante la infancia (3).
Los casos de deficiencia adquirida del factor V se asocian con deficiencias de los factores VII, X y II y se encuentran en hepatopatías (cirrosis, hepatitis, falla o insuficiencia hepática) por lo cual, al tener una vida media mucho más corta que la albúmina puede ser de mucha utilidad como marcador de la capacidad sintética del hígado en casos de enfermedades hepáticas agudas. Otras patologías que podrían dar déficit del factor V son la coagulación intravascular diseminada (CID), leucemia mielocítica crónica, policitemia vera, leucemia mielocítica aguda y falla renal crónica entre otras. También se pueden adquirir anticuerpos que interfieran con el factor V después de dar a luz, después de una cirugía o a través de ciertas enfermedades autoinmunes.
Los síntomas de la deficiencia de factor V son generalmente leves. Algunas personas podrían no presentar ningún síntoma, otras, como los homocigotas que presentan déficit congénito, los síntomas comienzan a temprana edad durante la niñez (4).
Entre los síntomas más comunes podemos mencionar:
• hemorragias nasales (epistaxis).
• propensión a moretones.
• períodos menstruales abundantes o prolongados (menorragia).
• hemorragias en la boca, después de cirugías o extracciones dentales.
• Otros síntomas reportados:
• hemorragias gastrointestinales.
• hemorragias musculares.
• hemorragias articulares (hemartrosis).
La deficiencia de factor V se diagnostica mediante una serie de pruebas sanguíneas:
– Pruebas de coagulación: TP y KPTT
– Análisis del factor V: la prueba consiste en medir el tiempo de coagulación de la muestra en un sistema que contiene todos los factores de coagulación de modo constante y en exceso (suministrado por el reactivo), con excepción del factor V que es aportado por la muestra analizada.
En la personas con concentraciones anormales de factor V también deberían verificarse las concentraciones del factor VIII a fin de descartar la posibilidad de una deficiencia combinada de factor V y factor VIII, lo que constituye un trastorno totalmente diferente.
El tratamiento para la deficiencia (congénita) de factor V generalmente solo se requiere en casos de hemorragias graves o antes de cirugías. El plasma fresco congelado (PFC) es el tratamiento usual porque no existe un concentrado exclusivamente de factor V. Las transfusiones de plaquetas, que contienen factor V en sus gránulos a, también constituyen una opción en algunos casos (5).
Prestaciones disponibles en Cibic:
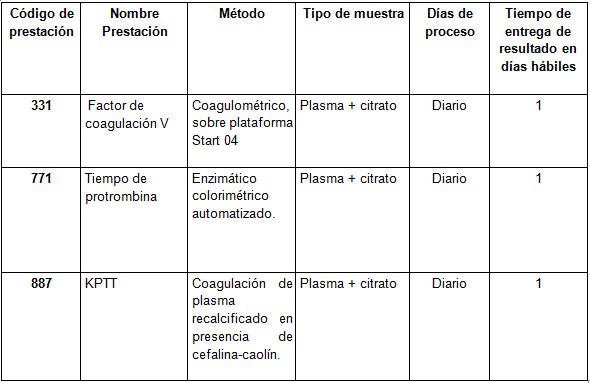
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Bibliografía:
1- Segers K, Dahback B, Nicolaes G. Coagulation factor V and trombophilia: background and mechanisms. Thrombosis Haemost 2007; 98(3): 530-542
2- Bolton-Maggs PH, Perry D, Chalmers EA. The rare coagulations disorders- review with guidelines for management from UKHCDO. Haemophilia 2004; 10: 593-628.
3- Mansouritorgabeh H, Rezaieyazdi Z, Pourfathollah AA, Rezai J, Esamaili H. Haemorrahagic symptoms in patients with combined factor V and VIII deficiency in north-eastern Iran. Haemophilia 2004; 10(3): 271-275.
4- United Kingdom Haemophilia Center Doctors Organisation. Guidelines on the selection and use of therapeutic products to treat haemophilia and other hereditary bleeding disorders. Haemophilia 2003; 9:1-23.
5- Ragni MV. Hemorrhagic disorders: coagulation factor deficiencies. In: Goldman L, Schafer Al, eds. Cecil Medicine. 24th ed. Philadelphia, Pa: Saunders Elsevier; 2011: chap 167
Para mayor información o consultas:
Sección: Bioquímica
Bioq. Hernán Brescia
Tel: 0341- 4499444 Int: 215