
La Parálisis Periódica Hipocaliémica (hypoPP, por su abreviatura en inglés) es una enfermedad hereditaria poco frecuente que pertenece al grupo de las canalopatías. Consiste en la presentación de episodios ocasionales de parálisis muscular progresivos en intensidad y frecuencia acompañada de hipocaliemia. Se estima una prevalencia de alrededor de 1 entre 100.000 individuos (1).
La enfermedad normalmente aparece en la segunda década de la vida. La hypoPP se transmite como una enfermedad autosómica dominante con penetrancia incompleta, especialmente en mujeres (1). Aproximadamente un 55-77% de los casos está asociado con mutaciones en el gen CACNA1S del canal de calcio del músculo y un 10-20% de los casos está vinculado a mutaciones en el gen SCN4A del canal de sodio del músculo (2,3).
El diagnóstico de hypoPP se basa en historia de episodios de parálisis fláccida acompañada de niveles séricos bajo de potasio durante episodios (<0.9 to 3.0 mmol/L), – pero no entre los mismos; ausencia de miotonía, tanto en sus manifestaciones clínicas como en electromiografías; ausencia de hipertiroidismo, rasgos dismórficos y arritmias cardíacas; y presencia de historia familiar consistente con herencia autosómica dominante. De la totalidad de los casos que cumplen con el criterio diagnóstico para hypoPP, aproximadamente 55-77% presentan mutaciones en el gen CACNA1S y 10-20% en el gen SCN4A (2,3).
HypoPP tipo 1: en la actualidad son cinco las mutaciones en el gen CACNA1S (MIM*114208) más frecuentemente asociadas a hypoPP (tipo 1): p.Arg528Gly, p.Arg528His (exón 11); p.Arg897Ser (exón 21); p.Arg1239Gly y p.Arg1239His (exón 30) (2)
HypoPP tipo 2: Las mutaciones más frecuentemente asociadas a hypoPP tipo 2 en SCN4A (MIM*603967) se localizan en el exón 12 (p.Arg669His, p.Arg672Ser, p.Arg672His, p.Arg672Gly, p.Arg672Cys) y en el exón 18 ( p.Arg1132Gln) (3).
La mayoría de los casos afectados por hypoPP poseen un progenitor afectado por la misma patología; en algunos casos, puede observarse, como consecuencia de penetrancia incompleta, casos esporádicos sin ninguno de los progenitores afectados. Las mutaciones más frecuentemente asociadas a estos casos suelen ser p.Arg528His y p.Arg1239His en los exones 11 y 30, del gen CACNA1S, respectivamente (4).
Prestaciones disponibles en Cibic:
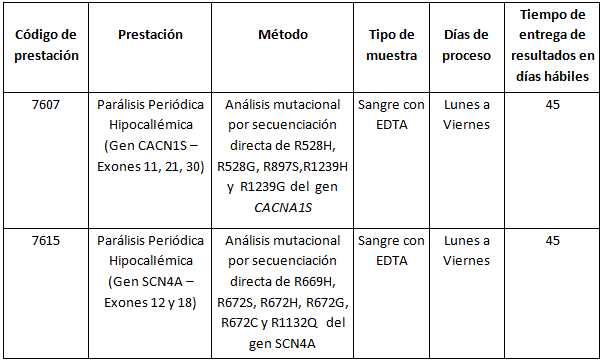
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Bibliografía
1. Sternberg D, et al. Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain. 2001;124:1091–9
2. Fontaine B, et al. Mapping of the hypokalaemic periodic paralysis (HypoPP) locus to chromosome 1q31-32 in three European families. Nature Genet. 1994;6: 267-72.
3. Bulman DE, et al. A novel sodium channel mutation in a family with hypokalemic periodic paralysis. Neurology. 1999;53:1932–6.
4. Kim SH, et al. Identification of mutations including de novo mutations in Korean patients with hypokalaemic periodic paralysis. Nephrol Dial Transplant. 2001;16:939–44.
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Analía Seravalle Tel: 0341 4499444 Int: 242
Dra. María Florencia Gosso Tel: 0341 4499444 Int: 258