
Los neoplasmas mieloproliferativos comprenden un grupo heterogéneo de trastornos hematológicos crónicos los cuales se caracterizan por un aumento en la producción de uno o más linajes hematopoyéticos (granulocítico, eritroide o megacariocítico). La organización mundial de la salud incluye bajo esta denominación las siguientes patologías: leucemia mieloide crónica, leucemia neutrofílica crónica, leucemia eosinofílica crónica, mielofibrosis primaria, policitemia vera, trombocitemia esencial, mastocitosis y neoplasmas mieloproliferativos no clasificables (1). La heterogeneidad fenotípica de estas enfermedades se debe a la presencia de diferentes reordenamientos genéticos o mutaciones que se asocian con la proliferación clonal (2). El descubrimiento de estas alteraciones moleculares ha permitido su empleo con fines diagnósticos y de monitoreo terapéutico.
Leucemia mieloide crónica
Al diagnóstico, más del 95% de los casos presenta la translocación t(9;22)(q34;q11.2) que da origen al cromosoma Philadelphia, producto de la fusión de los genes ABL y BCR, localizados en los cromosomas 9 y 22 respectivamente. La proteína de fusión resultante más frecuente (95%) tiene un peso molecular de 210 KDa (p210), existiendo también un producto alternativo de 190 KDa (p190) presente en una frecuencia mucho menor (2%). Estas proteínas son las responsables de la transformación maligna ya que tienen una actividad tirosina quinasa desregulada (3).
Leucemia neutrofílica crónica
En esta patología, también se observa la translocación cromosómica t(9;22)(q34;q11.2), pero el producto resultante es una proteína de 230 KDa (p230) (4).
Leucemia eosinofílica crónica
Si bien no existe un marcador molecular específico, existen mutaciones en genes que codifican para receptores celulares que producen su hiperactividad y consecuentemente una proliferación celular aumentada. El rearreglo molecular más frecuente en este caso es el FIP1L1-PDGFR (5).
Policitemia vera
El desarrollo de esta patología se atribuye, en el 95% de los casos, a una mutación somática en el exón 14 del gen JAK2, lo que desencadena la auto-fosforilación constitutiva de las vías mediadas por las proteínas JAK-STAT. Esta mutación conlleva al cambio de una valina por una fenilalanina en el codón 617 de la proteína (JAK2 V617F). Existen otras mutaciones (mutaciones puntuales, inserciones, deleciones), ubicadas en el exón 12 de JAK2 que pueden encontrarse en aquellos pacientes JAK2 V617F negativos y que tienen el mismo efecto patogénico (1).
Trombocitemia esencial
La mutación JAK2 V617F se encuentra en el 40-50% de los casos de trombocitemia esencial. En un porcentaje mucho menor (1-4%), se ha encontrado una mutación somática en el gen MPL que resulta en la sustitución de un triptofano por una leucina en el codón 515 de la proteína (MPL W515K/L) (4).
Mielofibrosis primaria
Desde el punto de vista molecular, esta patología no se ha asociado con ningún marcador, sin embargo, en el 50% de los casos se ha encontrado la mutación JAK2 V617F y se ha observado una correlación entre la carga mutacional al momento del diagnóstico y durante la progresión de la enfermedad, siendo un biomarcador útil como estratificador de riesgo. Por otro lado, la mutación MPL W515K/L se ha observado en un 4% de los casos de mielofibrosis primaria (6).
Mastocitosis
El 95% de los casos presenta una mutación somática puntual en el gen KIT que ocasiona el cambio de una valina por un aspartato en el codón 816 (KIT D816V) y consecuentemente una activación constitutiva de la actividad tirosina quinasa de la proteína para la cual codifica, desregulando de esta forma la proliferación celular (7).
Determinaciones disponibles en Cibic:
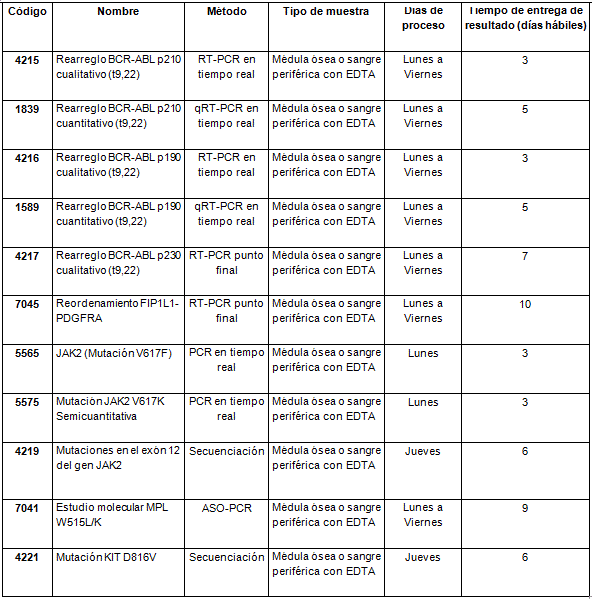
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones.
Bibliografía
1. Tefferi A and Vardiman J. Classification and diagnosis of myeloproliferative neoplasms: The 2008 World Health Organizationcriteria and point-of-care diagnostic algorithms. Leukemia. 2008;22:14-22.
2. Tefferi A, Gilliland DG. Oncogenes in myeloproliferative disorders. Cell Cycle. 2007;6:550-566.
3. Vardiman J, Melo J, Baccarani M, Thiele J. Chronic myelogenous leukaemia, BCR-ABL 1 positive. Lyon. Swerdlow S, Campo E, Harris N, Jaffe E, Pileri S, Stein H 2008;pp:32-37.
4. Pane F, Frigeri F, et al. Neutrophilic-chronic myeloid leukemia. Blood. 1996;88:2410-2414
5. Bain B, Gilliland D, Vardiman J. and Horny H. Chronic eosinophilic leukaemia not otherwise specified. Lyon. Swerdlow S, Campo E, Harris N, Jaffe E, Pileri S, Stein H 2008;pp:48-50.
6. Philip A, Peter J, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. 2008;112:141-149.
7. Cem A. Molecular diagnosis of mast cell disorders. Journal of molecular diagnostics. 2006;8:412-419.
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Analía Seravalle
Tel: 0341-4499444. Interno: 242