

La antitrombina III (AT) es un inhibidor fisiológico del sistema de coagulación perteneciente a la familia de las serpinas (inhibidores de serinproteasas). Es una glicoproteína de cadena simple codificada por un gen del cromosoma 1. Se sintetiza en el hígado, su concentración plasmática es de 112 a 140 mg/l y su vida media es de 2,5 días. Se presenta en dos isoformas que difieren en el grado de glicosilación: la isoforma α que representa el 90-95% de la AT en circulación y la isoforma no glicosilada β que se une a los glicosaminoglicanos de la pared vascular (1).
La función regulatoria de AT sobre el sistema de coagulación está ejercida principalmente por la inhibición de trombina (FIIa), pero también inhibe factor Xa, factor IXa, factor XIIa, factor XIIa, calicreinas, plasmina y el complejo FVIIa-FT. El mecanismo de inhibición de AT sobre la proteasa blanco es de trampa ya que la proteasa actúa proteolíticamente sobre el sitio reactivo de AT, lo cual produce un cambio conformacional y la proteasa queda unida covalentemente al inhibidor. Ésta es una inhibición progresiva y se acelera 500 veces por la presencia de heparina o glicosaminoglicanos, como el heparán sulfato de la superficie de las células endoteliales. Esta rápida inactivación de trombina y FXa en presencia de heparina es referida como actividad de cofactor de la heparina de la AT.
La deficiencia de AT está asociada con riesgo incrementado de trombosis venosa profunda, tromboembolismo pulmonar, trombosis arterial y pude ser hereditaria o adquirida.
Las deficiencias se clasifican de tipo I o cuantitativas y tipo II o cualitativas. En las deficiencias de tipo I tanto la actividad funcional como la concentración antigénica están igualmente disminuidas, sugiriendo un defecto en la síntesis o secreción de la proteína. En las de tipo II el defecto puede involucrar el sitio reactivo (tipo II RS), el sitio de unión a la heparina ( tipo II HBS) o estar presente más de un defecto funcional conocido como pleiotrópico (tipo II PE) (2).
El déficit hereditario se estima en una persona por cada 2000 a 5000, se trata de un desorden de carácter autosómico dominante que afecta a ambos sexos por igual. En los sujetos afectados que muestran unos niveles plasmáticos de AT de un 40 – 60 % por debajo de lo normal, aparecen episodios espontáneos de trombosis y embolia pulmonar, usualmente después de los 20 años, aumentando el riesgo con otros factores como la cirugía, los traumatismos y el embarazo. La frecuencia de episodios tromboembólicos durante el embarazo en pacientes con deficiencia hereditaria de AT es de hasta el 70%. Tener en cuenta que la mayoría de las deficiencias hereditarias son de tipo I heterocigota y el homocigota es incompatible con la vida (3).
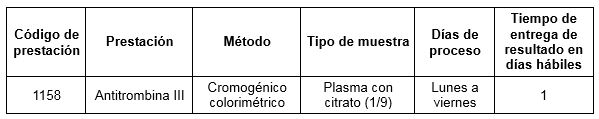
Prestación disponible en Cibic:
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Bibliografía
1- Rossi E, Pieroni G. Inhibidores fisiológicos de la coagulación, antitrombina. Fundamentos para el manejo práctico en el laboratorio de hemostasia. 2º edicion. Grupo Cooperativo Argentino de Hemostasia y Trombosis. Cap9: 476-482.2015
2- Muszbek L, Bereczky Z, Kovacs B. Antithrombin deficiency and its laboratory diagnosis. Clin Chem Lab Med. 48:67-78. 2010
3- Solanich T, Peñas C, Parriego V, Giménez A. Trombosis venosa profunda en gestantes con déficit de antitrombina III. Rev Angiología. Vol 62 núm 05:35-41. 2013
4- Ungerstedt JS, Schulman S, Egberg N. Discrepancy between antitrombin activity methods revealed in antithrombin stockholm: do factor Xa-based methods overstimate antithrombin activity in some patients. Blood vol 99:2271-2272.2009
Para mayor información o consultas:
Sección: Producción Bioquímica Clínica Automatizada
Bioq. Hernán G. Brescia
Tel: 0341-4499444. Int: 215