
{kind=link}
El Síndrome de Marfan (MIM #154700) es un desorden sistémico del tejido conectivo que presenta una amplia variabilidad clínica (1). Las manifestaciones fundamentales de esta enfermedad afectan a los sistemas oculares (miopía, desprendimiento de retina, cataratas, glaucoma), esquelético (talla alta, miembros desproporcionadamente largos, laxitud articular y deformidad de columna) y cardiovascular (dilatación de la raiz aórtica, prolapso de las válvulas mitral y tricúspide y dilatación de la arteria pulmonar proximal) (2,3).
Esta enfermedad sigue un patrón de herencia autosómico dominante, con alta penetrancia y marcada heterogeneidad fenotípica. En aproximadamente 70-93% de los casos se debe a mutaciones puntuales en el gen FBN1. Hasta en un 30% de los casos con resultado negativo para mutaciones en el gen FBN1 pueden identificarse grandes deleciones y/o duplicaciones (3).
El síndrome de Marfan-Like incluye múltiples patologías del tejido conectivo con manifestaciones clínicas que se superponen con el síndrome de Marfan, (ej. Síndrome de Loeys-Dietz, Enfermedad de Ehlers Danlos, Aneurisma aórtico torácico familiar). Los genes responsables de estas patologías son incluídos para realizar un correcto diagnóstico diferencial: ACTA2, CBS, COL3A1, COL5A1, COL5A2, FBN2, MYH11, SLC2A10, SMAD3, TGFBR1 ,TGFBR2 , TGFB2 y TGFB3(4).
En la actualidad, la tecnología de secuenciación de segunda generación (Next Generation Sequencing, NGS) permite realizar en forma simultánea el estudio de diferentes genes candidatos, facilitando y reduciendo el tiempo diangóstico, agilizando de esta forma la toma de decisiones en cuanto a la terapéutica y adecuado seguimiento del paciente y familiares en riesgo de portar mutaciones asociadas a estas patologías.
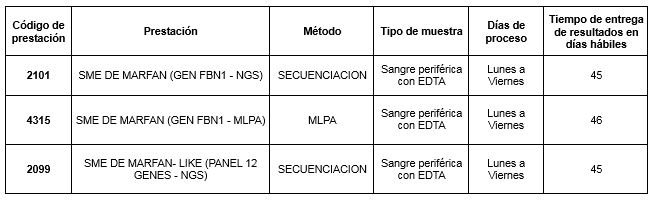
Prestaciones disponibles en Cibic:
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones o a la intranet.
Bibliografia
1. http://www.omim.org/
2. Barriales-Villa R y cols. Genética del Síndrome de Marfan. Cardiocore. 2011;46:101–104.
3. Cabrera Bueno F y cols.Nuevos criterios diagnósticos en el síndrome de Marfan. Cardiocore. 2011;46:85–88.
4. Fortuny E y cols. Aneurisma aórtico en síndromes hereditarios. Diagnóstico diferencial con el síndrome de Marfan. Cardiocore. 2011;46:105–108
Para mayor información o consultas:
Sección: Biología Molecular
Dra. Ma. Fernanda Madeira. Tel: 0341-4499444 Int: 241
Lic. Guadalupe Méjico.Tel: 0341-4499444 Int: 243
Bioq. María Florencia Gosso (PhD): Tel: 0341-4499444 Int: 258