
El síndrome de Alagille (ALGS, por sus siglas en inglés), conocido también como síndrome de Alagille-Watson y displasia arteriohepática, es una enfermedad multisistémica de herencia autosómica dominante de expresión variable tradicionalmente relacionada a un número reducido de conductos biliares en el hígado. ALGS se encuentra asociado a 5 características clínicas principales: colestasis, enfermedad cardíaca, anormalidades óseas, oculares, y rasgos faciales característicos (1). La prevalencia estimada en la población es de 1:30.000 nacidos.
ALGS es una enfermedad ocasionada por defectos en la vía de señalización del receptor Notch. Mutaciones puntuales en el gen JAG1 localizado en el brazo corto del cromosoma 20 (20p12.2), son causales de las alteraciones observadas en más del 90% de los pacientes con síndrome de Alagille tipo 1 (ALGS1). En más de la mitad de los casos se trata de mutaciones de novo. En alrededor de un 7% de pacientes con ALGS1 se han identificado deleciones de material genético en la región del cromosoma 20 que incluyen al gen JAG1 (2). Un pequeño porcentaje (<1%) es causado por mutaciones en el gen NOTCH2, asociándose a malformaciones renales (Síndrome de Alagille tipo 2, ALSG2) (3). EL gen JAG1 participa en procesos de señalización mediados por el receptor Notch esenciales para el correcto ensamblaje celular, originando alteraciones en el desarrollo y función tisular durante el desarrollo embrionario. Las manifestaciones clínicas asociadas a ALSG1 se observan generalmente al momento del nacimiento o en la infancia. La severidad de las manifestaciones clínicas varía entre individuos afectados, inclusive dentro de la misma familia (expresión variable), observándose en un amplio espectro de ellas, desde enfermedades cardíacas, renales o hepáticas graves hasta solo expresiones subclínicas (4).
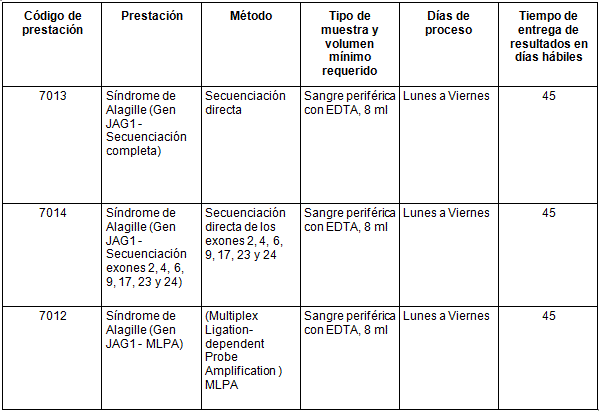
Determinaciones disponibles en CIBIC:
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Bibliografía
1. Li L, Krantz ID, Deng Y, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nature Genet. 1997; 16: 243-251.
2. Anad F, Burn J, Matthews D, et al. Alagille syndrome and deletion of 20p. J Med Genet. 1990; 27: 729-37.
3. McDaniell R, Warthen DM, Sanchez-Lara P A, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the Notch signaling pathway. Am. J. Hum. Genet. 2006; 79: 169-173.
4. Crosnier C, Lykavieris P, Meunier-Rotival M, Hadchouel M. Alagille syndrome: the widening spectrum of arteriohepatic dysplasia. Clinics in Liver Disease 2000; 4:765-78.
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Analía Seravalle Tel: 0341 4499444 Int: 242
Dra. María Florencia Gosso Tel: 0341 4499444 Int: 258