
En las enfermedades genéticas con mecanismos de herencia tradicional, se asume que los alelos procedentes de cada uno de los padres de un gen determinado, se expresan de manera idéntica en el hijo (herencia mendeliana). En el caso de la impronta genómica, los alelos son modificados en forma reversible en los gametos de manera que en el hijo los dos alelos se expresan en forma funcionalmente diferente. En otras palabras el alelo con impronta es heredado en forma mendeliana pero su expresión está determinada, a través de mecanismos epigenéticos, por el sexo del progenitor que trasmite el gen. Los ejemplos clásicos de estos mecanismos de herencia son los Síndromes de Angelman (AS) y Prader-Willi (PWS) los que son considerados el paradigma de la impronta genómica y de la disomía uniparental (1).
Síndrome de Angelman (AS). AS (MIM: #105830) es una enfermedad neurogénica compleja caracterizada por microcefalia, retraso severo del desarrollo, ausencia de lenguaje, marcha atáxica y/o temblor de extremidades. Posee un fenotipo conductual característico manifestado por episodios de risa espasmódica e inmotivada, hiperactividad y marcada inatención. La epilepsia es una manifestación prominente en el SA, estimándose que alrededor de un 6% de los individuos con retraso mental severo y epilepsia son portadores de este cuadro. AS afecta aproximadamente 1 en 15000 nacidos vivos (2). A nivel molecular, AS puede ser causado por diversos mecanismos: (i) deleción de la región AS/PWS en la región 15q11.2q1 de origen materno, (ii) disomía uniparenteral (DUP) de origen paterno, en la cual el padre aporta dos copias del cromosoma 15, (iii) defecto en el imprinting genómico, o (iv) mutación en el gen UBE3A, localizado en la región 15q11.2q13. El diagnóstico molecular inicial para AS consiste en el estudio de metilación de la región 15q11.2q13. Este estudio sirve para confirmar aproximadamente 80% de los casos de AS (patrón de metilación anormal). Posterior a la confirmación del diagnóstico, es posible la identificación del mecanismo molecular de AS por detección de deleciones y DUP mediante estudio de microsatélites, responsables de aproximadamente 70% y 7% de los casos, respectivamente (3). En un número reducido de casos, AS se presenta con patrones de metilación normales (11%). Ante este escenario, el estudio que se indica es la secuenciación del gen UBE3A. La importancia en la identificación del mecanismo molecular de AS es fundamental para calcular el riesgo relativo de un individuo a padecer y transmitirlo (riesgo de recurrencia en sucesivas generaciones).
Síndrome de Prader-Willi (PWS). PWS (MIM: #176270) es una enfermedad neurogénica que se caracteriza por hipotonía severa y dificultades para la alimentación en el período de recién nacido, seguido en el período preescolar por hiperfagia y obesidad mórbida secundaria. Todos los individuos tienen algún grado de retraso del desarrollo y una conducta impulsiva característica. Hombres y mujeres presentan hipogonadismo y la talla baja es frecuente. PWS afecta aproximadamente 1 en 25000 nacimientos vivos (1). PWS puede ser causado por uno de los siguientes mecanismos: (i) deleción de la región AS/PWS en la región 15q11.2q1 de origen paterno, (ii) disomía uniparenteral (DUP) de origen materno, en la cual la madre aporta dos copias del cromosoma 15, o (iii) defecto en el imprinting genómico (3).
El estudio de metilación de la región 15q11.2q13 es la técnica de elección para el diagnóstico molecular de PWS. Mediante esta sola técnica es posible diagnosticar PWS debido a cualquiera de los tres mecanismos previamente mencionados. Este estudio sirve para confirmar más de 99% de los casos de PWS (patrón de metilación anormal). Para investigar mecanismos de deleción y DUP materna en PWS, responsables del 75% y 20% de los casos, respectivamente, se realiza en estudio de microsatélites (3).
La importancia en la identificación del mecanismo molecular de PWS es fundamental para calcular el riesgo relativo de un individuo a padecer y transmitirlo (riesgo de recurrencia en sucesivas generaciones).
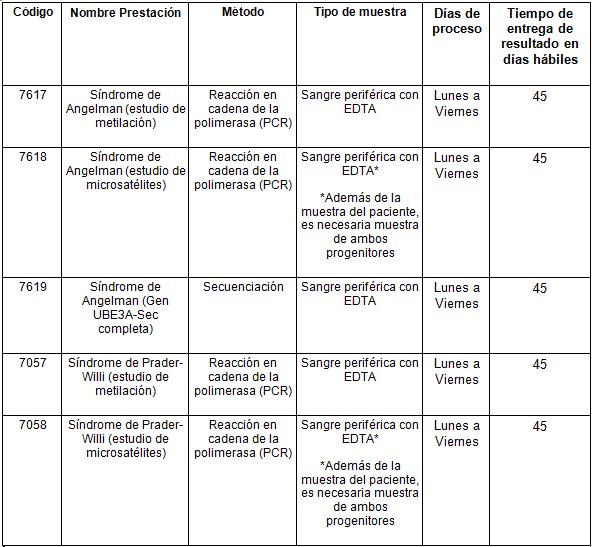
Determinaciones disponibles en Cibic:
Bibliografía
1. Cassidy SB y cols. Prader-Willi and Angelman syndromes: disorders of genomic imprinting. Medicine 1998; 77: 140-151.
2 Williams CA, y cols. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet A. 2006; 140:413-8.
3. American Society of Human Genetics/American College of Medical Genetics Test and Technology Transfer Committee. Diagnostic testing for Prader-Willi and Angelman syndromes. Am. J. Hum. Genet.1996; 58: 1085-88.
Para mayor información o consultas:
Sección: Biología Molecular
Lic. Analía Seravalle Tel: 0341 4499444 Int: 242
Dra. María Florencia Gosso Tel: 0341 4499444 Int: 258