
La fibrosis quística (FQ), también llamada Mucoviscidosis o enfermedad fibroquística, es una enfermedad autosómica recesiva y es causada por mutaciones en la molécula del regulador de la conductancia transmembrana de la fibrosis quística, lo que resulta en un defecto de la secreción de cloruro por las células. Si bien afecta diversas glándulas, lo que produce síntomas a nivel gastrointestinal y pulmonar, éstos últimos dominan el curso clínico. A nivel pulmonar existe clearence mucociliar reducido, alteración de la secreción mucosa, inflamación crónica e infecciones crónicas. Las células afectadas son las de los epitelios y está afectado el transporte de iones, con la consiguiente alteración del contenido de agua. Las secreciones son viscosas lo que provoca que un grupo de bacterias en particular suelen infectar a estos pacientes y además generando cepas mucoides y muchas veces resistentes a los agentes antimicrobianos. Los principales microorganismos que infectan a estos pacientes son Pseudomonas aeruginosa, Staphylococcus aureus, Burkholderia cepacia complex y Haemophilus influenzae, aunque también se puede presentar Streptococcus pneumoniae y Burkholderia multivorans. Otros bacilos gram negativos como Achromobacter xylosoxidans, Stenotrophomonas maltophilia y micobacterias no tuberculosis pueden aparecer en adultos. Los esfingolípidos, especialmente la ceramida, que causa inflamación y elevada susceptibilidad a las infecciones bacterianas juegan un papel crucial en la patogénesis. Los patógenos previamente colonizan la nasofaringe. No está demostrado el papel de los virus pero se ha propuesto la vacunación frente a virus Influenza. S aureus y P aeruginosa son los agentes primarios de la infección pulmonar de los pacientes con FQ, estando presente el primero en 50 a 60 % de los pacientes y pudiendo aislarse cepas meticilino resistentes (SAMR). P aeruginosa infecta hasta 70 % de los pacientes y las cepas van evolucionando desde lisas, resistentes al suero y serotipificables a rugosas, sensibles al suero y no tipificables, elaborando grandes cantidades de un polímero mucoide extracelular asociado con las células y formando una biopelícula. Las colonias se vuelven mucoides en los medios de cultivo habituales. P aeruginosa comienza siendo sensible a los antibacterianos beta lactámicos anti pseudomonas, las quinolonas y los aminoglucósidos pero va progresivamente adquiriendo resistencia por la presión selectiva de los tratamientos, quedando a veces sólo sensible a las polimixinas. Una evolución desfavorable permite la infección por B cepacia complex y otras especies de Burkholderia.
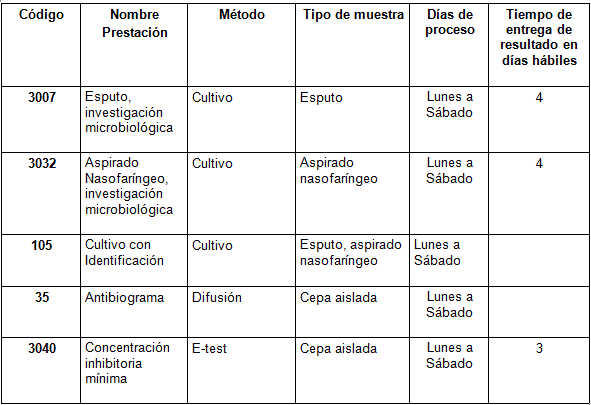
Determinaciones disponibles en Cibic:
Bibliografía
1- Grassmé H et al. Ceramide in cystic fibrosis. Handb Exp Pharmacol 2013; 216:265-74.
2- Verhagen LM et al. Bacterial respiratory pathogens in children with inherited immune and airway disorders: nasopharyngeal carriage and disease risk. Pediatr Infect Dis J. 2013; 32:399-404
3- Miller JK et al. Mathematical modelling of Pseudomonas aeruginosa biofilm growth and treatment in the cystic fibrosis lung. Math Med Biol 2013; en prensa.
4- Ciofu O et al. Respiratory bacterial infections in cystic fibrosis. Curr Opin Pulm Med 2013; 19:251-8
5- Stokell JR et al. Rapid emergence of a ceftazidime-resistant Burkholderia multivorans strain in a cystic fibrosis patient. J Cyst Fibros. 2013; pii:S1569-1993.
6- Lo DK et al. Interventions for the eradication of methicillin-resistant Staphylococcus aureus(MRSA) in people with cystic fibrosis. Cochrane Database Syst Rev 2013 en prensa.
Para mayor información o consultas:
Sección: Microbiología
Dra. Noemí Borda
Tel: (0341) 4499444 Int 228