
La galactosemia es una enfermedad rara autosómica recesiva caracterizada por niveles elevados de galactosa y sus metabolitos debido a deficiencia en las enzimas implicadas en su metabolismo. El metabolismo de galactosa a glucosa involucra la acción de tres enzimas: galactoquinasa (GALK), galactosa -1-fosfato uridil transferasa (GAT) y UDP – galactosa 4 – epimerasa (GALE). El tipo y la gravedad de la enfermedad dependen de la enzima alterada.
La galactosemia clásica (OMIM 230400), la forma más común y más grave de la enfermedad, es causada por la deficiencia de GALT, la enzima que cataliza el metabolismo de la galactosa-1-fosfato a uridina difosfato galactosa. La elevación resultante de gal-1-fosfato conduce a retraso intelectual disfunción hepática y el desarrollo de catarata. La incidencia de galactosemia clásica es 1:40000-60000 recién nacidos en poblaciones caucásicas. Los recién nacidos afectados con la variante clásica se comportan normalmente, sin embargo, después de la exposición a la leche, que contiene abundantes galactosa, desarrollan síntomas que se agravan rápidamente desde vómitos y diarrea a ictericia, retraso del crecimiento, hepatomegalia y sepsis por E. coli, que puede ser letal. El diagnóstico neonatal y la restricción inmediata de la galactosa de la dieta previenen o resuelven los síntomas agudos de la galactosemia clásica. Sin embargo, a pesar incluso del diagnóstico presintomático y la estricta intervención dietética de por vida, gran parte de los pacientes desarrollan complicaciones preocupantes a largo plazo. Las secuelas incluyen deterioro cognitivo y / o conductual en casi la mitad de los pacientes, dificultades del habla en al menos la mitad de los pacientes, baja densidad mineral ósea en muchos pacientes, ataxia o temblor en algunos pacientes, e insuficiencia ovárica primaria o prematura en al menos el 80 % de todas las niñas y mujeres.
Las mutaciones en el gen GALT pueden dar lugar al tipo clásico grave o a galactosemia Duarte, que es una condición leve prácticamente asintomática. Las mutaciones que provocan escasa o nula actividad de la enzima GALT se consideran mutaciones clásicas, mientras que las variantes Duarte reducen la actividad de la enzima en aproximadamente un 25 %. Hay numerosas mutaciones clásicas que se producen en todo el gen GALT. Una combinación de dos alelos clásicos provoca galactosemia clásica, mientras que un heterocigoto mixto de alelo clásico y un alelo variante Duarte, se asocia con la variante Duarte de galactosemia. El diagnóstico genético es útil para confirmar el diagnóstico de galactosemia y proveer información que puede conducir a un mejor manejo de la enfermedad. Además, el diagnóstico genético en los portadores es importante para el asesoramiento genético.
Prestación disponible en Cibic:
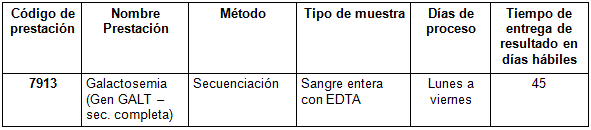
Para conocer las condiciones del paciente, de almacenamiento y de envío de la muestra y otros datos sobre las prácticas consulte al manual de prestaciones y a la extranet.
Bibliografía
1. Elsas, L.J. Galactosemia. In: Pagon RA, Bird TD, Dolan CR, et al., editors. GeneReviews. Seattle (WA): University of Washington, Seattle; 26 October 2010.
2. Fridovich-Keil J, Walter J (2008) Galactosemia. In: Valle D, Beaudet A, Vogelstein B, Kinzler K, Antonarakis S, and Ballabio A (eds). The online metabolic and molecular bases of inherited disease – OMMBID, chap. 72. www.ommbid.com.
3. Reichardt, J.K., Woo, S.L. Molecular basis of galactosemia: mutations and polymorphisms in the gene encoding human galactose-1-phosphate uridylyltransferase. Proceedings of the National Academy of Sciences. 1991; 88(7):2633–2637.
Para mayor información o consultas:
Sección: Biología Molecular
Dra. María Sonia Baquedano
Lic. Analía Seravalle
Tel: 0341-4499444. Interno: 241 – 242