
La Hiperplasia Adrenal Congénita (HAC) es una enfermedad autosómica recesiva frecuente que en la mayoría de los casos (90%) es causada por la deficiencia de la enzima 21-hidroxilasa (P450c21) en la corteza adrenal. La deficiencia de esta enzima conduce a grados variables de alteración de la síntesis de cortisol y aldosterona acompañado por un aumento en la biosíntesis de andrógenos. Dependiendo de la actividad residual de la enzima, presenta tres formas clínicas. Dos de ellas se expresan fenotípicamente durante la vida fetal, y se denominan formas clásicas. Estas incluyen una forma severa, potencialmente letal si no se trata, en la cual está alterada tanto la síntesis de cortisol como de aldosterona, conocida como forma perdedora de sal; y una forma menos severa, en la cual la función mineralocorticoide esta conservada, conocida como forma virilizante simpleLa forma clásica de deficiencia de P450c21 es la causa más común de ambigüedad genital en las mujeres. Finalmente, ha sido descripta una forma de expresión tardía, en la cual el exceso de producción de andrógenos adrenales se manifiesta en la prepubertal o adolescencia con hirsutismo o infertilidad en las mujeres y se denomina forma no clásica. La frecuencia de las formas clásicas es 1/10000 a 1/18000 nacidos vivos, siendo la incidencia de portadores heterocigotas de 1 en 60. La forma no clásica es aún mucho más frecuente.
La enzima P450c21 se localiza en el retículo endoplásmico y cataliza la conversión de progesterona y 17-hidroxiprogesterona a 11-desoxicorticosterona y 11-desoxicortisol, respectivamente. En humanos, el gen de la P450c21 (CYP21B) está presente junto con un pseudogen no funcional (CYP21A). Estos genes están localizados dentro de la región génica del HLA clase II en el brazo corto del cromosoma 6. El pseudogen es 98% y 96% homólogo al gen activo en exones e intrones, respectivamente. Ambos genes tienen 3.1 Kb en longitud y están codificados por 10 exones. La mayoría de las mutaciones puntuales deletéreas residen en el pseudogen y son transferidas al gen activo por eventos de conversión génica, causando tanto inactivación completa de la enzima P450c21, como parcial. La alta homología y la complejidad de este locus génico provocan considerables dificultades en su análisis molecular requiriendo personal capacitado en la interpretación de los resultados.
Debido a su alta tasa de incidencia, las pruebas de deficiencia de P450c21 están incluidas en la mayoría de los programas de screening neonatal, normalmente mediante la medición de las concentraciones de 17-hidroxiprogesterona en manchas de sangre por inmunoensayo. Sin embargo, debido a las altas tasas de falsos positivos de los inmunoensayos (debido a elevaciones fisiológicas de 17-hidroxiprogesterona en bebés prematuros y a la reactividad cruzada del inmunoensayo con otros esteroides), se requiere la confirmación repitiendo el estudio después de varias semanas. En un pequeño porcentaje de casos, las pruebas adicionales no proporcionan un diagnóstico definitivo. Además, muchas veces no se detectan las formas no clásicas y se requiere la evaluación de un perfil hormonal más extenso incluyendo las pruebas antes y después de la estimulación suprarrenal con hormona corticotropina (ACTH).
Por estas razones, el diagnóstico genético juega un papel auxiliar importante en las formas clásicas, y es aún más importante en las formas no clásicas. Además, el diagnóstico genético es importante para el asesoramiento genético debido a la alta frecuencia de portadores de mutaciones en el gen de la P450c21 (CYP21B). Por último, la prueba genética puede jugar un papel como complemento de las pruebas bioquímicas de líquido amniótico en el diagnóstico prenatal de deficiencia de P450c21. Esta representa uno de los pocos trastornos para los que el diagnóstico prenatal y el tratamiento son factibles y eficaces para prevenir defectos de nacimiento.
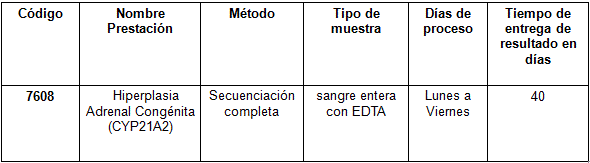
Determinación disponible en Cibic:
Bibliografía
1. Collett-Solberg PF: Congenital adrenal hyperplasias: from clinical genetics and biochemistry to clinical practice, part I. Clin Pediatr 40:1-16, 2001.
2. Mercke DP, Bornstein SR, Avila NA, Chrousos GP: NIH conference: future directions in the study and management of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Ann Intern Med 136:320-334, 2002.
3. Speiser PW, White PC: Medical progress: congenital adrenal hyperplasia. N Engl J Med 349:776-788, 2003.
4. Auchus RJ, Chang AY.46,XX DSD: the masculinised female. Best Pract Res Clin Endocrinol Metab 24:219-242, 2010.
Para mayor información o consultas:
Sección: Biología Molecular
Dra. María Sonia Baquedano
Lic. Analía Seravalle
Tel: 0341-4499444. Interno: 241 – 242